
New Approach for Detection of Normal Alternative Splicing Events and Aberrant Spliceogenic Transcripts with Long-Range PCR and Deep RNA Sequencing

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
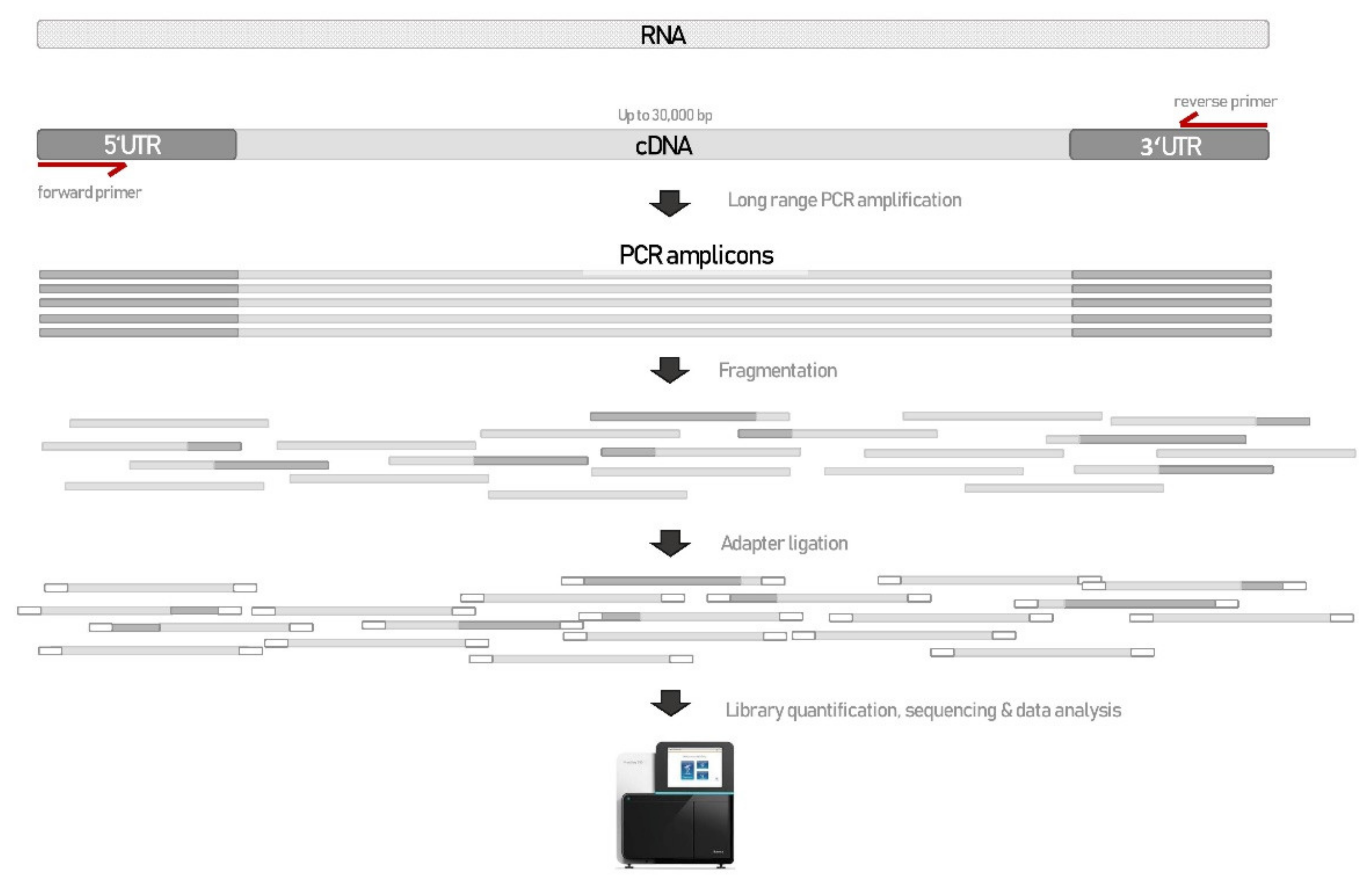
2. Materials and Methods
2.1. Patient Samples
2.2. DNA Sequencing—DNAseq
2.3. RNA Sequencing—RNAseq
2.4. Alternative Splicing Events Threshold
2.5. Identification of Splicing Aberrations Caused by Genetic Variant
3. Results
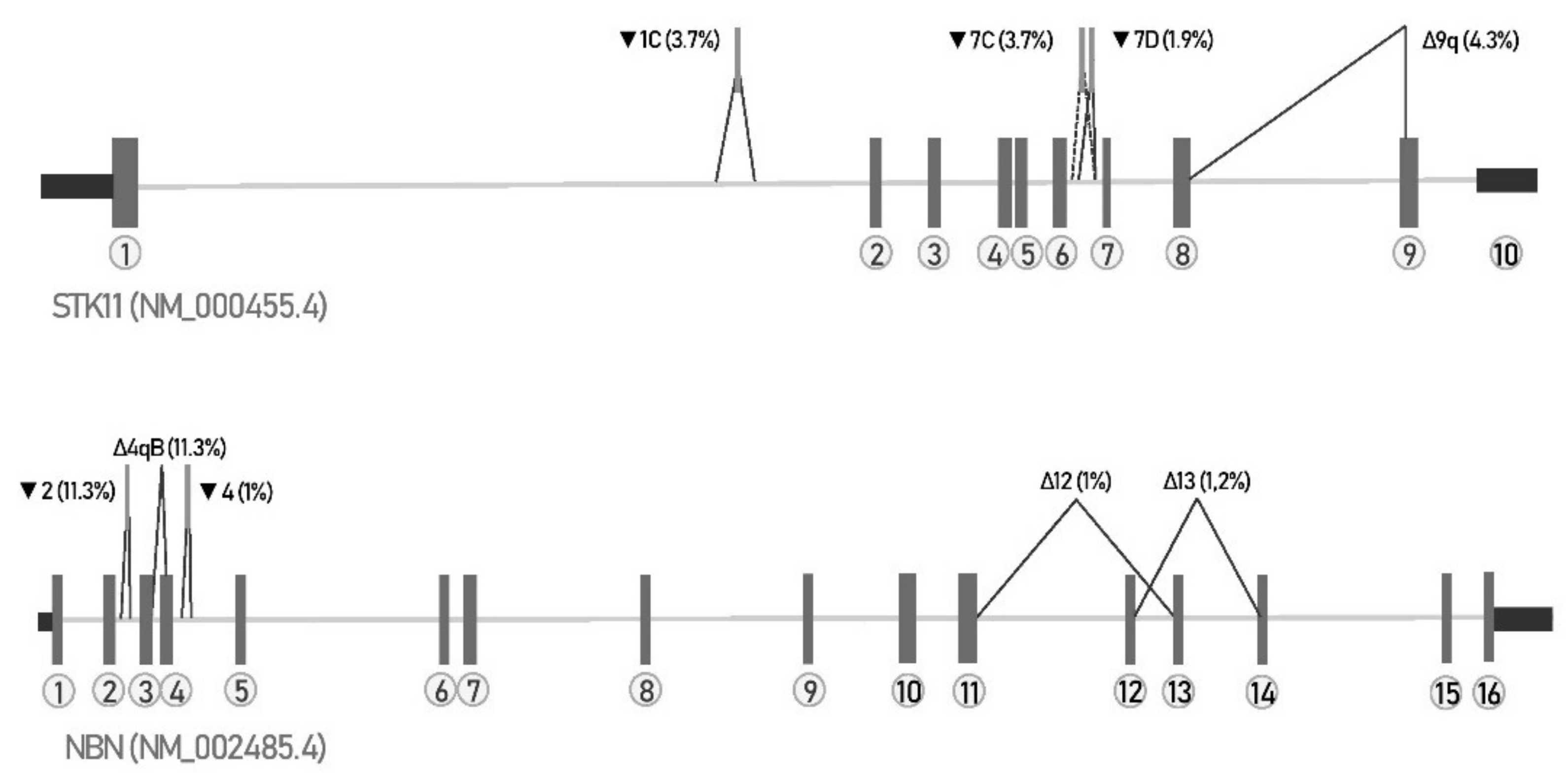
3.1. Method Confirmation—Alternative Splicing Events in STK11 Gene
3.2. Catalogue of Naturally Occurring Splicing Events in NBN Gene
3.3. Detection of Known Spliceogenic Variants
3.4. Determination of Spliceogenicity of VUS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Colombo, M.; Blok, M.J.; Whiley, P.; Santamariña, M.; Gutiérrez-Enríquez, S.; Romero, A.; Garre, P.; Becker, A.; Smith, L.D.; De Vecchi, G.; et al. Comprehensive annotation of splice junctions supports pervasive alternative splicing at the BRCA1 locus: A report from the ENIGMA consortium. Hum. Mol. Genet. 2014, 23, 3666–3680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, M.; Suzuki, H.; Shiraishi, Y.; Kosaki, K. Effectiveness of integrated interpretation of exome and corresponding transcriptome data for detecting splicing variants of genes associated with autosomal recessive disorders. Mol. Genet. Metab. Rep. 2019, 21, 100531. [Google Scholar] [CrossRef] [PubMed]
- Cartegni, L.; Chew, S.L.; Krainer, A.R. Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet. 2002, 3, 285–298. [Google Scholar] [CrossRef]
- Karam, R.; Conner, B.; LaDuca, H.; McGoldrick, K.; Krempely, K.; Richardson, M.E.; Zimmermann, H.; Gutierrez, S.; Reineke, P.; Hoang, L.; et al. Assessment of Diagnostic Outcomes of RNA Genetic Testing for Hereditary Cancer. JAMA Netw. Open 2019, 2, e1913900. [Google Scholar] [CrossRef] [Green Version]
- Whiley, P.J.; De La Hoya, M.; Thomassen, M.; Becker, A.; Brandão, R.; Pedersen, I.S.; Montagna, M.; Menéndez, M.; Quiles, F.; Gutiérrez-Enríquez, S.; et al. Comparison of mRNA splicing assay protocols across multiple laboratories: Recommendations for best practice in standardized clinical testing. Clin. Chem. 2014, 60, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, P.; Frommolt, P. Challenges in the Setup of Large-scale Next-Generation Sequencing Analysis Workflows. Comput. Struct. Biotechnol. J. 2017, 15, 471–477. [Google Scholar] [CrossRef]
- Ozsolak, F.; Milos, P.M. RNA sequencing: Advances, challenges and opportunities. Nat. Rev. Genet. 2011, 12, 87–98. [Google Scholar] [CrossRef]
- Davy, G.; Rousselin, A.; Goardon, N.; Castéra, L.; Harter, V.; Legros, A.; Muller, E.; Fouillet, R.; Brault, B.; Smirnova, A.S.; et al. Detecting splicing patterns in genes involved in hereditary breast and ovarian cancer. Eur. J. Hum. Genet. 2017, 25, 1147–1154. [Google Scholar] [CrossRef] [Green Version]
- Brandão, R.D.; Mensaert, K.; López-Perolio, I.; Tserpelis, D.; Xenakis, M.; Lattimore, V.; Walker, L.C.; Kvist, A.; Vega, A.; Gutiérrez-Enríquez, S.; et al. Targeted RNA-seq successfully identifies normal and pathogenic splicing events in breast/ovarian cancer susceptibility and Lynch syndrome genes. Int. J. Cancer 2019, 145, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Setrajcic Dragos, V.; Blatnik, A.; Klancar, G.; Stegel, V.; Krajc, M.; Blatnik, O.; Novakovic, S. Two novel NF1 pathogenic variants causing the creation of a new splice site in patients with neurofibromatosis type I. Front. Genet. 2019, 10, 762. [Google Scholar] [CrossRef] [Green Version]
- Klančar, G.; Blatnik, A.; Šetrajčič Dragoš, V.; Vogrič, V.; Stegel, V.; Blatnik, O.; Drev, P.; Gazič, B.; Krajc, M.; Novaković, S. A Novel Germline MLH1 In-Frame Deletion in a Slovenian Lynch Syndrome Family Associated with Uncommon Isolated PMS2 Loss in Tumor Tissue. Genes (Basel) 2020, 11, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Gingeras, T.R. Mapping RNA-seq Reads with STAR. Curr. Protoc. Bioinform. 2015, 51, 11–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varon, R.; Dutrannoy, V.; Weikert, G.; Tanzarella, C.; Antoccia, A.; Stöckl, L.; Spadoni, E.; Krüger, L.-A.; di Masi, A.; Sperling, K.; et al. Mild Nijmegen breakage syndrome phenotype due to alternative splicing. Hum. Mol. Genet. 2006, 15, 679–689. [Google Scholar] [CrossRef] [Green Version]
- Tessitore, A.; Biordi, L.; Flati, V.; Toniato, E.; Marchetti, P.; Ricevuto, E.; Ficorella, C.; Scotto, L.; Giannini, G.; Frati, L.; et al. New mutations and protein variants ofNBS1 are identified in cancer cell lines. Genes Chromosom. Cancer 2003, 36, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Takakuwa, T.; Luo, W.-J.; Francisca Ham, M.; Aozasa, K. A 50-bp insertion from intron 2 between exons 2 and 3 ofNBS1 may be a spliced variant. Genes, Chromosom. Cancer 2004, 39, 341–342. [Google Scholar] [CrossRef]
- Mayr, C. Evolution and Biological Roles of Alternative 3′UTRs. Trends Cell Biol. 2016, 26, 227–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrith, T.; Li, B.; Cass, A.A.; Conner, B.R.; LaDuca, H.; McKenna, D.B.; Maxwell, K.N.; Domchek, S.; Morman, N.A.; Heinlen, C.; et al. Splicing profile by capture RNA-seq identifies pathogenic germline variants in tumor suppressor genes. Npj Precis. Oncol. 2020, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Farber-Katz, S.; Hsuan, V.; Wu, S.; Landrith, T.; Vuong, H.; Xu, D.; Li, B.; Hoo, J.; Lam, S.; Nashed, S.; et al. Quantitative Analysis of BRCA1 and BRCA2 Germline Splicing Variants Using a Novel RNA-Massively Parallel Sequencing Assay. Front. Oncol. 2018, 8, 286. [Google Scholar] [CrossRef] [Green Version]
- Casadei, S.; Gulsuner, S.; Shirts, B.H.; Mandell, J.B.; Kortbawi, H.M.; Norquist, B.S.; Swisher, E.M.; Lee, M.K.; Goldberg, Y.; O’Connor, R.; et al. Characterization of splice-altering mutations in inherited predisposition to cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 26798–26807. [Google Scholar] [CrossRef]
- Krivokuca, A.; Dragos, V.S.; Stamatovic, L.; Blatnik, A.; Boljevic, I.; Stegel, V.; Rakobradovic, J.; Skerl, P.; Jovandic, S.; Krajc, M.; et al. Novel BRCA1 splice-site mutation in ovarian cancer patients of Slavic origin. Fam. Cancer 2018, 17, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Q.; Chen, J.-H.; Chen, Y.-C.; Chen, M.-Y.; Hsieh, C.-Y.; Teng, S.-C.; Wu, K.-J. Interaction between NBS1 and the mTOR/Rictor/SIN1 Complex through Specific Domains. PLoS ONE 2013, 8, e65586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houdayer, C.; Caux-Moncoutier, V.; Krieger, S.; Barrois, M.; Bonnet, F.; Bourdon, V.; Bronner, M.; Buisson, M.; Coulet, F.; Gaildrat, P.; et al. Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Hum. Mutat. 2012, 33, 1228–1238. [Google Scholar] [CrossRef]
- Kukurba, K.R.; Montgomery, S.B. RNA Sequencing and Analysis. Cold Spring Harb. Protoc. 2015, 2015, 951–969. [Google Scholar] [CrossRef] [Green Version]
- Hrdlickova, R.; Toloue, M.; Tian, B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA 2017, 8, e1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| RNA Consequence | Junction Description | Average Number of Reads Supporting the Junction (N = 6) | Mean Percentage of Junction Reads † | Biotype | Percentage of Samples with Observed Junction | Detected by Brandão et al., 2019 |
|---|---|---|---|---|---|---|
| r.-272_-186del | Δ5′UTR | 64 | 0.026 | terminal modification | 33 | no |
| r.-245_-209del | Δ5′UTR | 56 | 0.023 | terminal modification | 50 | no |
| r.-258_-185del | Δ5′UTR | 150 | 0.061 | terminal modification | 50 | no |
| r.-323_597del | Δ5′UTR | 86 | 0.029 | terminal modification | 33 | no |
| r.290_291ins290+2456_290+2554 | ▼1A | 608 | 0.207 | cryptic exon inclusion | 67 | yes |
| r.290_291ins290+5106_290+5326 | ▼1B | 44 | 0.015 | cryptic exon inclusion | 50 | yes |
| r.290_291ins291-2149_291-2019 | ▼1C | 10,912 | 3.717 | cryptic exon inclusion | 100 | yes |
| r.290_291ins291-2038_291-102 | ▼1D | 70 | 0.024 | cryptic exon inclusion | 83 | yes |
| r.290_291ins291-2897_291-2755 | ▼1E | 72 | 0.025 | cryptic exon inclusion | 33 | no |
| r.290_291ins291-2149_291-1782 | ▼1F | 105 | 0.071 | cryptic exon inclusion | 67 | no |
| ▼1G | 146 | 0.050 | cryptic exon inclusion | 100 | yes | |
| r.290_291ins290+114_290+190 ‡ | ▼1H | 116 | 0.040 | cryptic exon inclusion | 17 | yes |
| r.290_291ins291-2149_291-1324 ‡ | ▼1I | 175 | 0.060 | cryptic exon inclusion | 17 | yes |
| intron 1 junction | / | 293,569 | / | intron 1 junction | 100 | yes |
| r.374delinsAC | Δ2pA | 405 | 0.141 | exonic donor shift | 100 | no |
| r.374insA_375delG | Δ2pB | 117 | 0.041 | exonic donor shift | 100 | no |
| r.373_376del | Δ2,3q | 274 | 0.096 | exonic acceptor shift | 100 | no |
| r.291_464del | Δ2–3 | 1140 | 0.373 | multiple exon skipping | 100 | yes |
| intron 2 junction | / | 286,038 | / | intron 2 junction | 100 | yes |
| r.373_378del | Δ3q | 227 | 0.079 | exonic acceptor shift | 67 | no |
| intron 3 junction | / | 316,895 | / | intron 3 junction | 100 | yes |
| r.465_597del | Δ4 | 532 | 0.186 | exon skipping | 100 | yes |
| r.490_653del | Δ4p,Δ5q | 67 | 0.022 | mixed | 50 | no |
| r.465_734del § | Δ4–5 | 6 | 0.002 | multiple exon skipping | 100 | yes |
| r.465_920del | Δ4–7 | 62 | 0.023 | multiple exon skipping | 100 | yes |
| intron 4 junction | / | 254,161 | / | intron 4 junction | 100 | yes |
| r.706_734del | Δ5 | 342 | 0.125 | exon skipping | 83 | yes |
| r.734del | Δ5p | 29 | 0.010 | exonic donor shift | 83 | no |
| intron 5 junction | / | 295,148 | / | intron 5 junction | 100 | yes |
| r.862del | Δ6p | 36 | 0.015 | exonic donor shift | 100 | no |
| r.862_863ins862+281_863-103 | ▼7A | 1545 | 0.661 | cryptic exon inclusion | 100 | yes |
| r.862_863ins862+286_863-103 | ▼7B | 1218 | 0.521 | cryptic exon inclusion | 100 | yes |
| r.862_863ins863-283_863-103 | ▼7C | 3230 | 1.382 | cryptic exon inclusion | 100 | yes |
| r.862_863ins863-253_863-103 | ▼7D | 4570 | 1.956 | cryptic exon inclusion | 100 | yes |
| r.862_863ins863-195_863-103 | ▼7E | 186 | 0.079 | cryptic exon inclusion | 67 | no |
| r.858_862del+r.862_863ins863-125_863-103 | ▼7F | 16 | 0.007 | mixed | 33 | no |
| r.862_863ins863-126_863-103 | ▼7G | 25 | 0.011 | cryptic exon inclusion | 33 | no |
| r.862_863ins863-125_863-103 | ▼7H | 202 | 0.087 | cryptic exon inclusion | 100 | no |
| r.820_921ins921-34_921-1 ‡ | ▼7q | 29 | 0.012 | intronic acceptor shift | 17 | yes |
| intron 6 junction | / | 233,667 | / | intron 6 junction | 100 | yes |
| r.920_921ins921-105_921-1 | ▼8qA | 185 | 0.074 | intronic acceptor shift | 100 | yes |
| r.920_921ins921-87_921-1 | ▼8qB | 186 | 0.074 | intronic acceptor shift | 83 | yes |
| intron 7 junction | / | 250,858 | / | intron 7 junction | 100 | yes |
| r.1180_1181ins1108+466_1108+600 | ▼8A | 749 | 0.331 | cryptic exon inclusion | 100 | yes |
| r.1180_1181ins_1180+1_1108+187 | ▼8p | 141 | 0.062 | intronic donor shift | 33 | no |
| r.1180_1181ins1108+466_1109-641 | ▼8B | 95 | 0.042 | cryptic exon inclusion | 50 | yes |
| intron 8 junction | / | 226,042 | / | intron 8 junction | 100 | yes |
| r.1109_1113del | Δ9q | 9881 | 4.371 | exonic acceptor shift | 100 | yes |
| r.1109_*16del | Δ9 | 67 | 0.047 | exon skipping | 33 | yes |
| intron 9 junction | / | 56,173 | / | intron 9 junction | 100 | yes |
| RNA Consequence | Junction Description | Average Number of Reads Supporting the Junction (N = 6) | Mean Percentage of Junction Reads † | Biotype | Percentage of Samples with Observed Junction | Detected by Varon et al., 2006 |
|---|---|---|---|---|---|---|
| r.37_38ins37+466_37+648 | ▼1A | 72 | 0.179 | cryptic exon inclusion | 50 | No |
| r.37_38ins37+698_37+779 | ▼1B | 33 | 0.080 | cryptic exon inclusion | 50 | No |
| intron 1 junction | / | 52,217 | / | intron 1 junction | 100 | No |
| r.38_40del | Δ2q | 17 | 0.033 | exonic acceptor shift | 67 | No |
| r.171_172ins172-479_172-430 | ▼2 | 12,105 | 11.297 | cryptic exon inclusion | 100 | yes |
| r.171_172ins172-27_172-1 | ▼3q | 50 | 0.070 | intronic acceptor shift | 83 | No |
| r.171_172ins171+1_171+4 | ▼4p | 35 | 0.048 | intronic donor shift | 100 | No |
| intron 2 junction | / | 72,077 | / | intron 2 junction | 100 | No |
| r.38_171del | Δ2 | 31 | 0.169 | exon skipping | 83 | No |
| intron 3 junction | / | 115,981 | / | intron 3 junction | 100 | No |
| r.172_320del | Δ3–4 | 21 | 0.020 | multiple exon skipping | 33 | No |
| r.172_320del | Δ3 | 137 | 0.145 | exon skipping | 83 | No |
| r.321_325del | Δ4qA | 37 | 0.032 | exonic acceptor shift | 67 | No |
| r.321_361del | Δ4qB | 3341 | 2.881 | exonic acceptor shift | 100 | No |
| r.172_361del NM_001024688.2 | 31 | / | exon skipping+exonic acceptor shift | 83 | No | |
| r.172_361del | Δ3+4qC | 118 | 0.126 | exon skipping+exonic acceptor shift | 67 | No |
| r.480_481ins480+306_480+395 | ▼4 | 1294 | 1.041 | cryptic exon inclusion | 100 | No |
| intron 4 junction | / | 138,006 | / | intron 4 junction | 100 | No |
| r.38_480del | Δ2–4 | 24 | 0.023 | multiple exon skipping | 83 | No |
| r.321_480del | Δ4 | 58 | 0.045 | exon skipping | 50 | No |
| r.172_480del | Δ3–4 | 43 | 0.040 | multiple exon skipping | 33 | No |
| r.481del | Δ5qB | 22 | 0.016 | exonic acceptor shift | 100 | No |
| intron 5 junction | / | 135,653 | / | intron 5 junction | 100 | No |
| r.481_584del | Δ5 | 891 | 0.651 | exon skipping | 100 | Yes |
| r.321_584del | Δ4–5 | 161 | 0.116 | multiple exon skipping | 100 | Yes |
| r.172_584del | Δ3–5 | 37 | 0.035 | multiple exon skipping | 33 | No |
| r.38_584del | Δ2–5 | 164 | 0.174 | multiple exon skipping | 83 | No |
| r.589del | Δ6qB | 86 | 0.064 | exonic acceptor shift | 100 | No |
| intron 6 junction | / | 139,974 | / | intron 6 junction | 100 | No |
| r.585_702del | Δ6 | 75 | 0.054 | exon skipping | 50 | No |
| r.703_820del | Δ7q | 127 | 0.090 | exonic acceptor shift | 50 | Yes |
| intron 7 junction | / | 179,463 | / | intron 7 junction | 100 | No |
| r.585_896del | Δ6–7 | 118 | 0.075 | multiple exon skipping | 83 | Yes |
| r.481_896del | Δ5–7 | 202 | 0.224 | multiple exon skipping | 67 | No |
| r.38_896del | Δ2–7 | 61 | 0.052 | multiple exon skipping | 67 | No |
| r.897del | Δ8q | 17 | 0.010 | exonic acceptor shift | 100 | No |
| r.994_995ins994+1178_995-1769 | ▼8A | 86 | 0.041 | cryptic exon inclusion | 67 | No |
| r.994_995ins995-1769_995-1604 | ▼8B | 34 | 0.015 | cryptic exon inclusion | 33 | No |
| intron 8 junction | / | 202,305 | / | intron 8 junction | 100 | No |
| r.1124_1125ins1124+703_1124+760 | ▼9 | 391 | 0.220 | cryptic exon inclusion | 100 | Yes |
| intron 9 junction | / | 181,550 | / | intron 9 junction | 100 | No |
| r.995_1124del | Δ9 | 173 | 0.270 | exon skipping | 100 | No |
| intron 10 junction | / | 127,561 | / | intron 10 junction | 100 | No |
| r.1398del | Δ11qA | 29 | 0.023 | exonic acceptor shift | 100 | No |
| r.1398_1403del | Δ11qB | 83 | 0.065 | exonic acceptor shift | 33 | No |
| r.1398_1471del | Δ11qC | 70 | 0.055 | exonic acceptor shift | 83 | No |
| r.1845_1846ins1845+1521_1845+1597 | ▼11 | 22 | 0.030 | cryptic exon inclusion | 33 | No |
| r.1845_1846ins1846-23_1846-1 | ▼12q | 68 | 0.052 | intronic acceptor shift | 83 | No |
| intron 11 junction | / | 132,009 | / | intron 11 junction | 100 | No |
| r.1846_1849del | Δ12q | 46 | 0.035 | exonic acceptor shift | 67 | No |
| intron 12 junction | / | 128,083 | / | intron 12 junction | 100 | No |
| r.1896_1914del | Δ12p | 152 | 0.118 | exonic donor shift | 100 | No |
| r.1846_1914del | Δ12 | 894 | 1.030 | exon skipping | 100 | No |
| r.1915_1932del | Δ13qA | 316 | 0.247 | exonic acceptor shift | 100 | No |
| r.1915_2009del | Δ13qB | 114 | 0.089 | exonic acceptor shift | 67 | Yes |
| intron 13 junction | / | 128,636 | / | intron 13 junction | 100 | No |
| r.1915_2070del | Δ13 | 1576 | 1.228 | exon skipping | 100 | Yes |
| r.1846_2070del | Δ12–13 | 96 | 0.074 | multiple exon skipping | 83 | No |
| r.2184_2185ins2184+417_2184+464 | ▼14A | 59 | 0.042 | cryptic exon inclusion | 100 | No |
| r.2184_2185ins2184+1511_2184+1578 | ▼14B | 58 | 0.036 | cryptic exon inclusion | 67 | No |
| r.2184_2185ins2185-735_2185-610 | ▼14C | 76 | 0.059 | cryptic exon inclusion | 83 | No |
| r.2184_2185ins2185-718_2185-610 | ▼14D | 184 | 0.149 | cryptic exon inclusion | 100 | Yes |
| r.2184_2185ins2185-4_2185-1ins | ▼14q | 33 | 0.026 | intronic acceptor shift | 33 | No |
| intron 14 junction | / | 123,705 | / | intron 14 junction | 100 | No |
| r.2071_2184del | Δ14 | 98 | 0.077 | exon skipping | 100 | No |
| r.1915_2184del | Δ13–14 | 45 | 0.073 | multiple exon skipping | 83 | No |
| r.1846_2184del | Δ12–14 | 222 | 0.174 | multiple exon skipping | 100 | Yes |
| intron 15 junction | / | 136,201 | / | intron 15 junction | 100 | No |
| r.2185_2234del | Δ15 | 70 | 0.054 | exon skipping | 83 | No |
| r.1915_2234del | Δ13–15 | 100 | 0.075 | multiple exon skipping | 100 | No |
| r.*39_*541del | Δ3′UTR | 93 | 0.068 | terminal modification | 67 | No |
| r.2003_*1085del | Δ13p+Δ14–15 | 74 | 0.058 | multiple exon skipping+exonic donor shift | 33 | No |
| r.*1076_*1143del | Δ3′UTR | 27 | 0.020 | terminal modification | 33 | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dragoš, V.Š.; Stegel, V.; Blatnik, A.; Klančar, G.; Krajc, M.; Novaković, S. New Approach for Detection of Normal Alternative Splicing Events and Aberrant Spliceogenic Transcripts with Long-Range PCR and Deep RNA Sequencing. Biology 2021, 10, 706. https://doi.org/10.3390/biology10080706
Dragoš VŠ, Stegel V, Blatnik A, Klančar G, Krajc M, Novaković S. New Approach for Detection of Normal Alternative Splicing Events and Aberrant Spliceogenic Transcripts with Long-Range PCR and Deep RNA Sequencing. Biology. 2021; 10(8):706. https://doi.org/10.3390/biology10080706
Chicago/Turabian StyleDragoš, Vita Šetrajčič, Vida Stegel, Ana Blatnik, Gašper Klančar, Mateja Krajc, and Srdjan Novaković. 2021. "New Approach for Detection of Normal Alternative Splicing Events and Aberrant Spliceogenic Transcripts with Long-Range PCR and Deep RNA Sequencing" Biology 10, no. 8: 706. https://doi.org/10.3390/biology10080706