Post-Polymerization Modification of Poly(L-glutamic acid) with D-(+)-Glucosamine

Abstract
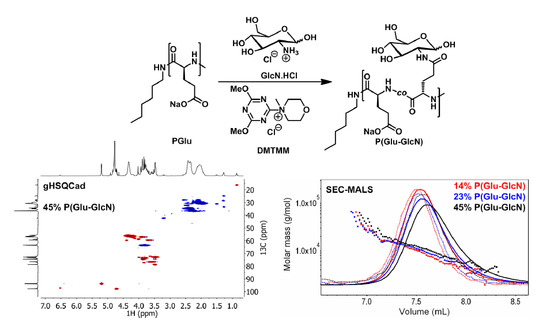
:
1. Introduction
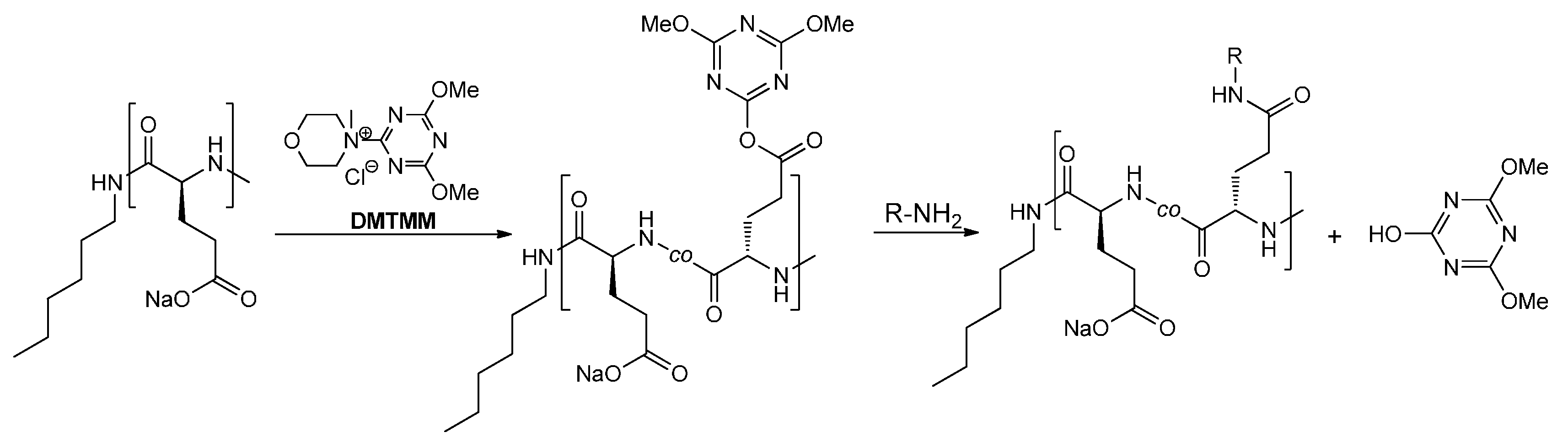
2. Results and Discussion







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
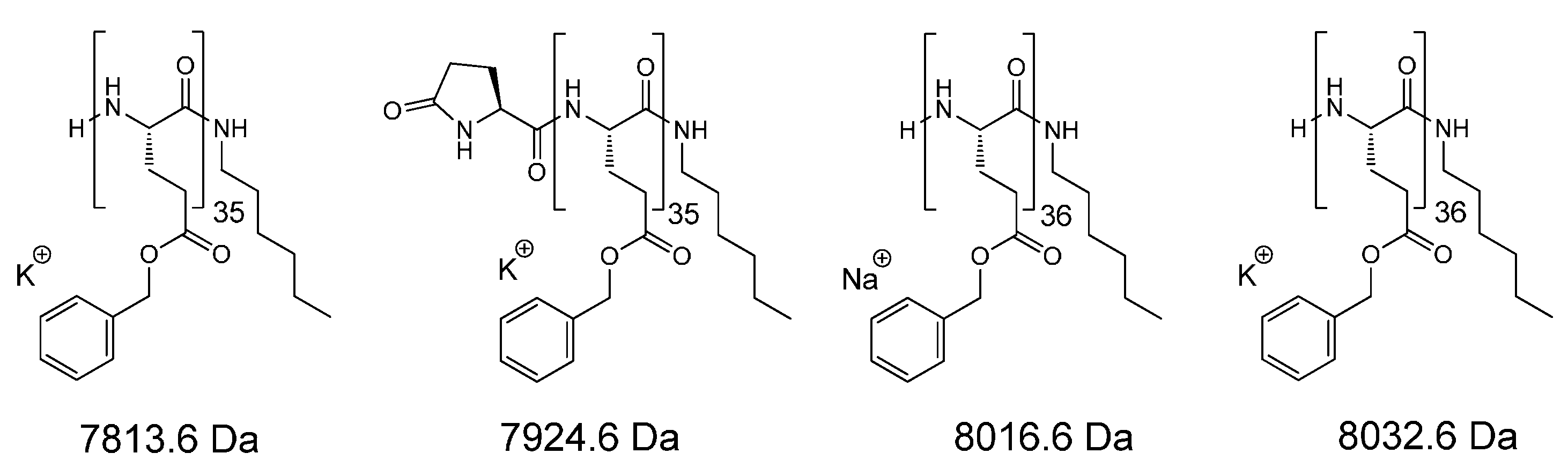
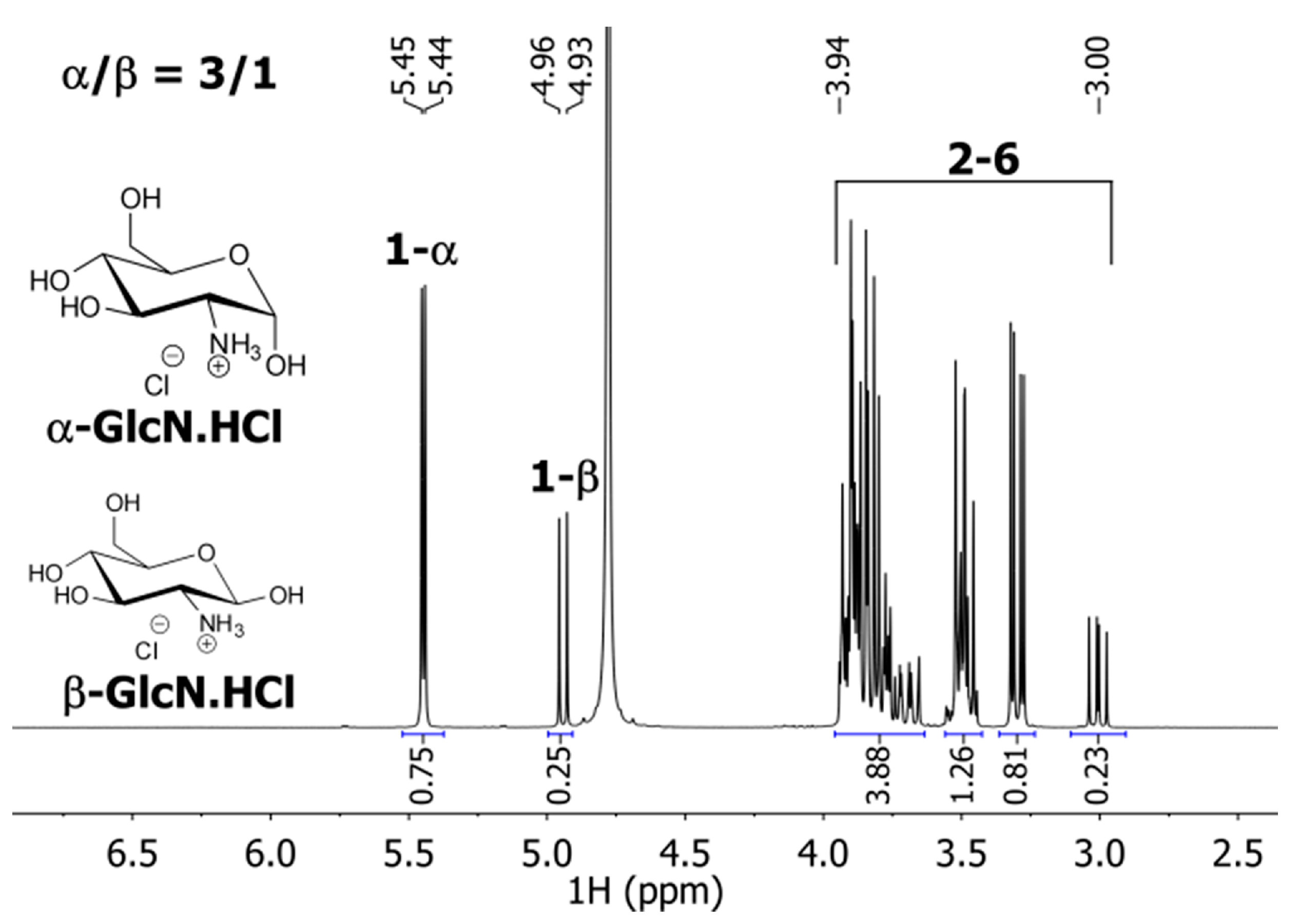
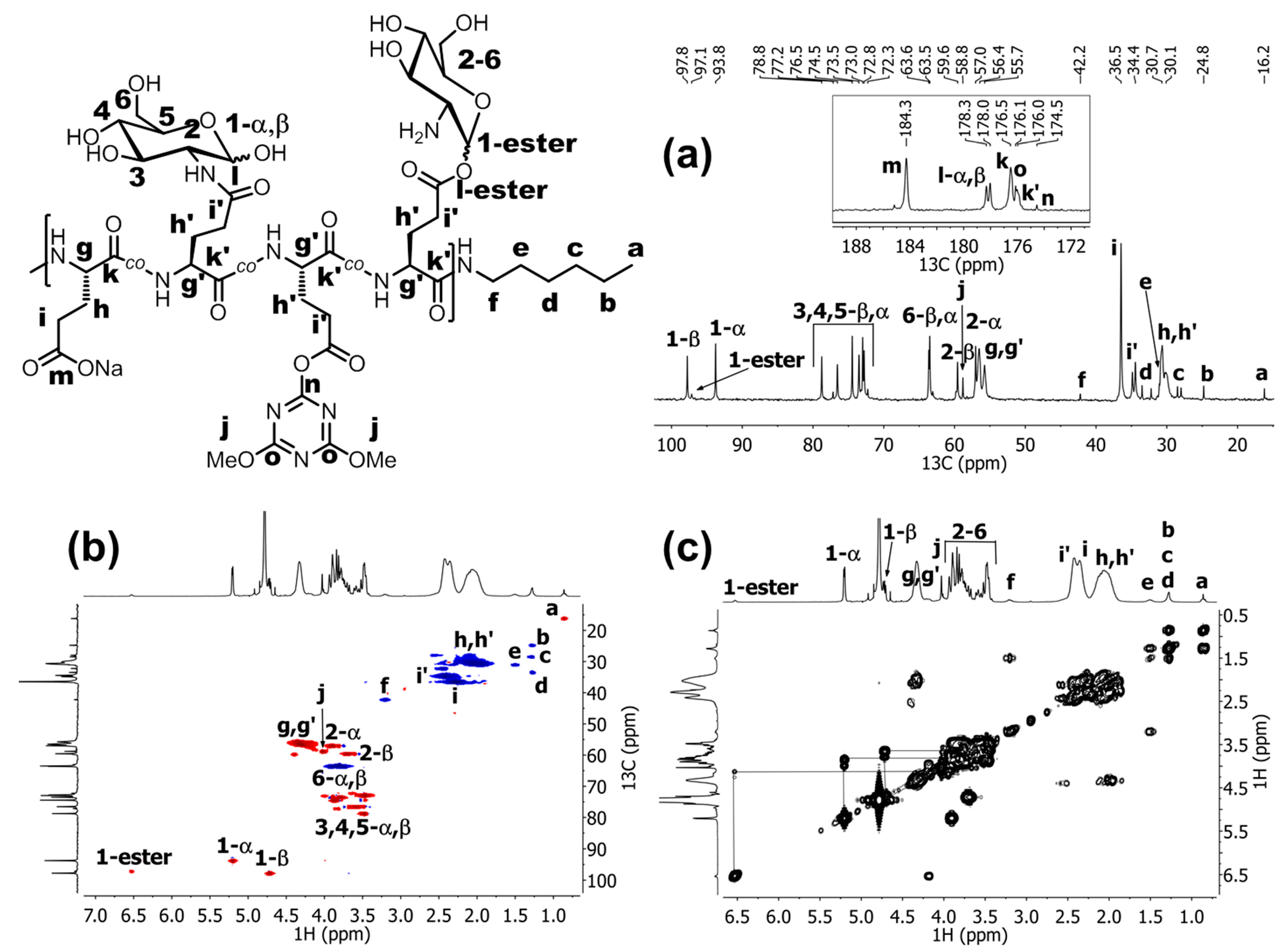{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Denotation | Reaction Conditions | 1H-NMR | (Mn)calc. (kDa) | SEC MALS | Yield (%) | |||
|---|---|---|---|---|---|---|---|---|
| Glu:GlcN:DMTMM | DG (%) | DS(triazinyl) (%) | Mn (kDa) | Mw (kDa) | ĐM | |||
| PGlu | - | - | - | - | 6.4 | 6.9 | 1.1 | 95 |
| 14% P(Glu-GlcN) | 1:0.5:0.25 | 14 | 1 | 7.5 | 7.4 | 9.2 | 1.2 | 87 |
| 23% P(Glu-GlcN) | 1:1:0.5 | 23 | 2 | 8.1 | 8.0 | 9.5 | 1.2 | 74 |
| 45% P(Glu-GlcN) | 1:1.5:0.75 | 45 | 3 | 9.3 | 8.9 | 9.9 | 1.1 | 76 |




3. Experimental Section
3.1. Materials
3.2. Methods
3.2.1. NMR
3.2.2. Size-Exclusion Chromatography Coupled to a Multi-Angle Light-Scattering Detector (SEC-MALS)
3.2.3. Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF MS)
3.3. Synthesis
3.3.1. γ-Benzyl-L-glutamate NCA (BGlu NCA)
3.3.2. Poly(γ-benzyl-L-glutamate) (PBGlu)
3.3.3. Poly(sodium-L-glutamate) (PGlu)
3.3.4. General Procedure for Modification of PGlu with GlcN (P(Glu-GlcN))
- 14% P(Glu-GlcN): PGlu (1 mmol Glu units), DMTMM (0.25 mmol), GlcN.HCl (0.5 mmol). Yield: 161 mg (87%). SEC-MALS: Mn = 7.4 kDa, Mw = 9.2 kDa, ĐM = 1.2.
- 23% P(Glu-GlcN): PGlu (1 mmol Glu units), DMTMM (0.50 mmol), GlcN.HCl (1.0 mmol). Yield: 164 mg (74%). SEC-MALS: Mn = 8.0 kDa, Mw = 9.5 kDa, ĐM = 1.2.
- 45% P(Glu-GlcN): PGlu (1 mmol Glu units), DMTMM (0.75 mmol), GlcN.HCl (1.5 mmol). Yield: 193 mg (76%). SEC-MALS: Mn = 8.9 kDa, Mw = 9.9 kDa, ĐM = 1.1.
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Spain, S.G.; Cameron, N.R. A spoonful of sugar: The application of glycopolymers in therapeutics. Polym. Chem. 2010, 2, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Dhaware, V.; Shaikh, A.Y.; Kar, M.; Hotha, S.; Sen Gupta, S. Synthesis and Self-assembly of Amphiphilic Homoglycopolypeptide. Langmuir 2013, 29, 5659–5667. [Google Scholar] [CrossRef] [PubMed]
- Spain, S.G.; Gibson, M.I.; Cameron, N.R. Recent advances in the synthesis of well-defined glycopolymers. J. Polym. Sci. Part Polym. Chem. 2007, 45, 2059–2072. [Google Scholar] [CrossRef]
- Choi, S.-K.; Mammen, M.; Whitesides, G.M. Generation and in Situ Evaluation of Libraries of Poly(acrylic acid) Presenting Sialosides as Side Chains as Polyvalent Inhibitors of Influenza-Mediated Hemagglutination. J. Am. Chem. Soc. 1997, 119, 4103–4111. [Google Scholar] [CrossRef]
- Parry, A.L.; Clemson, N.A.; Ellis, J.; Bernhard, S.S.R.; Davis, B.G.; Cameron, N.R. “Multicopy Multivalent” Glycopolymer-Stabilized Gold Nanoparticles as Potential Synthetic Cancer Vaccines. J. Am. Chem. Soc. 2013, 135, 9362–9365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashida, M.; Hirabayashi, H.; Nishikawa, M.; Takakura, Y. Targeted delivery of drugs and proteins to the liver via receptor-mediated endocytosis. J. Control. Release 1997, 46, 129–137. [Google Scholar] [CrossRef]
- Upadhyay, K.K.; Bhatt, A.N.; Mishra, A.K.; Dwarakanath, B.S.; Jain, S.; Schatz, C.; le Meins, J.-F.; Farooque, A.; Chandraiah, G.; Jain, A.K.; et al. The intracellular drug delivery and anti tumor activity of doxorubicin loaded poly(γ-benzyl l-glutamate)-b-hyaluronan polymersomes. Biomaterials 2010, 31, 2882–2892. [Google Scholar] [CrossRef] [PubMed]
- Erbacher, P.; Roche, A.C.; Monsigny, M.; Midoux, P. Glycosylated Polylysine/DNA Complexes: Gene Transfer Efficiency in Relation with the Size and the Sugar Substitution Level of Glycosylated Polylysines and with the Plasmid Size. Bioconjug. Chem. 1995, 6, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Peppas, N.A. Synthesis and characterization of pH-sensitive glycopolymers for oral drug delivery systems. J. Biomater. Sci. Polym. Ed. 2002, 13, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Borase, T.; Ninjbadgar, T.; Kapetanakis, A.; Roche, S.; O’Connor, R.; Kerskens, C.; Heise, A.; Brougham, D.F. Stable Aqueous Dispersions of Glycopeptide-Grafted Selectably Functionalized Magnetic Nanoparticles. Angew. Chem. Int. Ed. 2013, 52, 3164–3167. [Google Scholar] [CrossRef]
- Hayward, A.S.; Eissa, A.M.; Maltman, D.J.; Sano, N.; Przyborski, S.A.; Cameron, N.R. Galactose-Functionalized PolyHIPE Scaffolds for Use in Routine Three Dimensional Culture of Mammalian Hepatocytes. Biomacromolecules 2013, 14, 4271–4277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, H.-W.; Lai, J.-Y.; Tsai, W.-B. Galactosylated electrospun membranes for hepatocyte sandwich culture. Colloids Surf. B Biointerfaces 2014, 116, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.S.; Seo, S.J.; Park, I.K.; Kim, S.H.; Kim, T.H.; Hoshiba, T.; Harada, I.; Akaike, T. Galactose-carrying polymers as extracellular matrices for liver tissue engineering. Biomaterials 2006, 27, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Gibson, M.I.; Barker, C.A.; Spain, S.G.; Albertin, L.; Cameron, N.R. Inhibition of Ice Crystal Growth by Synthetic Glycopolymers: Implications for the Rational Design of Antifreeze Glycoprotein Mimics. Biomacromolecules 2009, 10, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Okada, M. Molecular design and syntheses of glycopolymers. Prog. Polym. Sci. 2001, 26, 67–104. [Google Scholar] [CrossRef]
- Davis, B.G. Synthesis of Glycoproteins. Chem. Rev. 2002, 102, 579–602. [Google Scholar] [CrossRef]
- Grande, D.; Baskaran, S.; Chaikof, E.L. Glycosaminoglycan Mimetic Biomaterials. 2. Alkene- and Acrylate-Derivatized Glycopolymers via Cyanoxyl-Mediated Free-Radical Polymerization. Macromolecules 2001, 34, 1640–1646. [Google Scholar] [CrossRef]
- Narain, R.; Armes, S.P. Synthesis of low polydispersity, controlled-structure sugar methacrylate polymers under mild conditions without protecting group chemistry. Chem. Commun. 2002, 2776–2777. [Google Scholar]
- Albertin, L.; Stenzel, M.H.; Barner-Kowollik, C.; Davis, T.P. Effect of an added base on (4-cyanopentanoic acid)-4-dithiobenzoate mediated RAFT polymerization in water. Polymer 2006, 47, 1011–1019. [Google Scholar] [CrossRef]
- Albertin, L.; Kohlert, C.; Stenzel, M.; Foster, L.J.R.; Davis, T.P. Chemoenzymatic Synthesis of Narrow-Polydispersity Glycopolymers: Poly(6-O-vinyladipoyl-d-glucopyranose). Biomacromolecules 2004, 5, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Collins, J.; Anastasaki, A.; Wallis, R.; Mitchell, D.A.; Becer, C.R.; Haddleton, D.M. Sequence-Controlled Multi-Block Glycopolymers to Inhibit DC-SIGN-gp120 Binding. Angew. Chem. Int. Ed. 2013, 52, 4435–4439. [Google Scholar] [CrossRef]
- Lu, H.; Wang, J.; Song, Z.; Yin, L.; Zhang, Y.; Tang, H.; Tu, C.; Lin, Y.; Cheng, J. Recent advances in amino acid N-carboxyanhydrides and synthetic polypeptides: Chemistry, self-assembly and biological applications. Chem. Commun. 2013, 50, 139–155. [Google Scholar] [CrossRef]
- Kramer, J.R.; Deming, T.J. Recent advances in glycopolypeptide synthesis. Polym. Chem. 2013, 5, 671–682. [Google Scholar] [CrossRef]
- Bonduelle, C.; Lecommandoux, S. Synthetic Glycopolypeptides as Biomimetic Analogues of Natural Glycoproteins. Biomacromolecules 2013, 14, 2973–2983. [Google Scholar] [CrossRef] [PubMed]
- Krannig, K.-S.; Schlaad, H. Emerging bioinspired polymers: Glycopolypeptides. Soft Matter 2014, 10, 4228–4235. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.R.; Deming, T.J. Glycopolypeptides via Living Polymerization of Glycosylated-l-lysine N-Carboxyanhydrides. J. Am. Chem. Soc. 2010, 132, 15068–15071. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.R.; Deming, T.J. Glycopolypeptides with a Redox-Triggered Helix-to-Coil Transition. J. Am. Chem. Soc. 2012, 134, 4112–4115. [Google Scholar] [CrossRef] [PubMed]
- Gibson, M.I.; Hunt, G.J.; Cameron, N.R. Improved synthesis of O-linked, and first synthesis of S- linked, carbohydrate functionalised N-carboxyanhydrides (glycoNCAs). Org. Biomol. Chem. 2007, 5, 2756–2757. [Google Scholar] [CrossRef] [PubMed]
- Pati, D.; Shaikh, A.Y.; Das, S.; Nareddy, P.K.; Swamy, M.J.; Hotha, S.; Gupta, S.S. Controlled Synthesis of O-Glycopolypeptide Polymers and Their Molecular Recognition by Lectins. Biomacromolecules 2012, 13, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Pati, D.; Shaikh, A.Y.; Hotha, S.; Gupta, S.S. Synthesis of glycopolypeptides by the ring opening polymerization of O-glycosylated-α-amino acid N-carboxyanhydride (NCA). Polym. Chem. 2011, 2, 805–811. [Google Scholar] [CrossRef]
- Stöhr, T.; Blaudszun, A.-R.; Steinfeld, U.; Wenz, G. Synthesis of glycosylated peptides by NCA polymerization for recognition of human T-cells. Polym. Chem. 2011, 2, 2239–2248. [Google Scholar] [CrossRef]
- Kramer, J.R.; Deming, T.J. Preparation of Multifunctional and Multireactive Polypeptides via Methionine Alkylation. Biomacromolecules 2012, 13, 1719–1723. [Google Scholar] [CrossRef] [PubMed]
- Gou, Y.; Geng, J.; Richards, S.-J.; Burns, J.; Remzi Becer, C.; Haddleton, D.M. A detailed study on understanding glycopolymer library and Con A interactions. J. Polym. Sci. Part Polym. Chem. 2013, 51, 2588–2597. [Google Scholar] [CrossRef]
- Richards, S.-J.; Jones, M.W.; Hunaban, M.; Haddleton, D.M.; Gibson, M.I. Probing Bacterial-Toxin Inhibition with Synthetic Glycopolymers Prepared by Tandem Post-Polymerization Modification: Role of Linker Length and Carbohydrate Density. Angew. Chem. Int. Ed. 2012, 51, 7812–7816. [Google Scholar] [CrossRef]
- Wu, P.; Malkoch, M.; Hunt, J.N.; Vestberg, R.; Kaltgrad, E.; Finn, M.G.; Fokin, V.V.; Sharpless, K.B.; Hawker, C.J. Multivalent, bifunctional dendrimers prepared by click chemistry. Chem. Commun. 2005, 5775–5777. [Google Scholar]
- Huang, J.; Habraken, G.; Audouin, F.; Heise, A. Hydrolytically Stable Bioactive Synthetic Glycopeptide Homo- and Copolymers by Combination of NCA Polymerization and Click Reaction. Macromolecules 2010, 43, 6050–6057. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, Q.; Li, G.-Z.; Haddleton, D.M.; Wallis, R.; Mitchell, D.; Heise, A.; Becer, C.R. Synthetic Glycopolypeptides as Potential Inhibitory Agents for Dendritic Cells and HIV-1 Trafficking. Macromol. Rapid Commun. 2013, 34, 1542–1546. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Bonduelle, C.; Thévenot, J.; Lecommandoux, S.; Heise, A. Biologically Active Polymersomes from Amphiphilic Glycopeptides. J. Am. Chem. Soc. 2012, 134, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Bonduelle, C.; Huang, J.; Ibarboure, E.; Heise, A.; Lecommandoux, S. Synthesis and self-assembly of “tree-like” amphiphilic glycopolypeptides. Chem. Commun. 2012, 48, 8353–8355. [Google Scholar] [CrossRef]
- Xiao, C.; Zhao, C.; He, P.; Tang, Z.; Chen, X.; Jing, X. Facile Synthesis of Glycopolypeptides by Combination of Ring-Opening Polymerization of an Alkyne-Substituted N-carboxyanhydride and Click “Glycosylation”. Macromol. Rapid Commun. 2010, 31, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Zhang, D. General Route toward Side-Chain-Functionalized α-Helical Polypeptides. Biomacromolecules 2010, 11, 1585–1592. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, A.J.; Deming, T.J. Soluble, Clickable Polypeptides from Azide-Containing N-Carboxyanhydride Monomers. ACS Macro Lett. 2013, 2, 351–354. [Google Scholar]
- Krannig, K.-S.; Schlaad, H. pH-Responsive Bioactive Glycopolypeptides with Enhanced Helicity and Solubility in Aqueous Solution. J. Am. Chem. Soc. 2012, 134, 18542–18545. [Google Scholar] [PubMed]
- Sun, J.; Schlaad, H. Thiol–Ene Clickable Polypeptides. Macromolecules 2010, 43, 4445–4448. [Google Scholar] [CrossRef]
- Robinson, J.W.; Schlaad, H. A versatile polypeptoid platform based on N-allyl glycine. Chem. Commun. 2012, 48, 7835–7837. [Google Scholar]
- Krannig, K.-S.; Huang, J.; Heise, A.; Schlaad, H. Photochemical thiol–yne functionalization of polypeptide scaffolds. Polym. Chem. 2013, 4, 3981–3986. [Google Scholar] [CrossRef]
- Tian, Z.; Wang, M.; Zhang, A.; Feng, Z. Study on synthesis of glycopeptide-based triblock copolymers and their aggregation behavior in water. Front. Mater. Sci. China 2007, 1, 162–167. [Google Scholar] [CrossRef]
- Hu, T.-C.; Korczyńska, J.; Smith, D.K.; Brzozowski, A.M. High-molecular-weight polymers for protein crystallization: Poly-γ-glutamic acid-based precipitants. Acta Crystallogr. Sect. D 2008, 64, 957–963. [Google Scholar] [CrossRef]
- Farkaš, P.; Čížová, A.; Bekešová, S.; Bystrický, S. Comparison of EDC and DMTMM efficiency in glycoconjugate preparation. Int. J. Biol. Macromol. 2013, 60, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Baek, M.-G. Glycodendrimers: Novel glycotope isosteres unmasking sugar coding. Case study with T-antigen markers from breast cancer MUC1 glycoprotein. Rev. Mol. Biotechnol. 2002, 90, 291–309. [Google Scholar] [CrossRef]
- Pelet, J.M.; Putnam, D. An In-Depth Analysis of Polymer-Analogous Conjugation Using DMTMM. Bioconjug. Chem. 2011, 22, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Mildner, R.; Menzel, H. Facile synthesis of pH-responsive glycopolypeptides with adjustable sugar density. J. Polym. Sci. Part Polym. Chem. 2013, 51, 3925–3931. [Google Scholar] [CrossRef]
- Bonduelle, C.; Mazzaferro, S.; Huang, J.; Lambert, O.; Heise, A.; Lecommandoux, S. Synthesis and self-assembly of branched glycopolypeptides: Effect of topology and conformation. Faraday Discuss. 2014, 166, 137–150. [Google Scholar] [CrossRef]
- Cameron, N.R.; Spain, S.G.; Kingham, J.A.; Weck, S.; Albertin, L.; Barker, C.A.; Battaglia, G.; Smart, T.; Blanazs, A. Synthesis of well-defined glycopolymers and some studies of their aqueous solution behaviour. Faraday Discuss. 2008, 139, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Eissa, A.M.; Smith, M.J.P.; Kubilis, A.; Mosely, J.A.; Cameron, N.R. Polymersome-forming amphiphilic glycosylated polymers: Synthesis and characterization. J. Polym. Sci. Part Polym. Chem. 2013, 51, 5184–5193. [Google Scholar] [CrossRef]
- Bui, L.; Abbou, S.; Ibarboure, E.; Guidolin, N.; Staedel, C.; Toulmé, J.-J.; Lecommandoux, S.; Schatz, C. Encapsidation of RNA–Polyelectrolyte Complexes with Amphiphilic Block Copolymers: Toward a New Self-Assembly Route. J. Am. Chem. Soc. 2012, 134, 20189–20196. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, K.K.; Meins, J.-F.L.; Misra, A.; Voisin, P.; Bouchaud, V.; Ibarboure, E.; Schatz, C.; Lecommandoux, S. Biomimetic Doxorubicin Loaded Polymersomes from Hyaluronan-block-Poly(γ-benzyl glutamate) Copolymers. Biomacromolecules 2009, 10, 2802–2808. [Google Scholar] [CrossRef] [PubMed]
- Schatz, C.; Louguet, S.; Le Meins, J.-F.; Lecommandoux, S. Polysaccharide-block-polypeptide Copolymer Vesicles: Towards Synthetic Viral Capsids. Angew. Chem. Int. Ed. 2009, 48, 2572–2575. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Mu, J.; Li, C. Synthesis and pH/sugar/salt-sensitivity study of boronate crosslinked glycopolymer nanoparticles. New J. Chem. 2013, 37, 796–803. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Han, Y.; Cheng, C.; Li, C. pH- and glucose-sensitive glycopolymer nanoparticles based on phenylboronic acid for triggered release of insulin. Carbohydr. Polym. 2012, 89, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Guo, Q.; Wu, Z.; Sun, L.; Zhang, Z.; Li, C.; Zhang, X. Amphiphilic glycopolymer nanoparticles as vehicles for nasal delivery of peptides and proteins. Eur. J. Pharm. Sci. 2013, 49, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Farkaš, P.; Bystrický, S. Efficient activation of carboxyl polysaccharides for the preparation of conjugates. Carbohydr. Polym. 2007, 68, 187–190. [Google Scholar] [CrossRef]
- Barz, M.; Duro-Castano, A.; Vicent, M.J. A versatile post-polymerization modification method for polyglutamic acid: Synthesis of orthogonal reactive polyglutamates and their use in “click chemistry”. Polym. Chem. 2013, 4, 2989–2994. [Google Scholar] [CrossRef]
- Thompson, K.; Michielsen, S. Novel synthesis of N-substituted polyacrylamides: Derivatization of poly(acrylic acid) with amines using a triazine-based condensing reagent. J. Polym. Sci. Part Polym. Chem. 2006, 44, 126–136. [Google Scholar] [CrossRef]
- Perdih, P.; Pahovnik, D.; Cegnar, M.; Miklavžin, A.; Kerč, J.; Žagar, E. Synthesis of chitosan-graft-poly(sodium-l-glutamate) for preparation of protein nanoparticles. Cellulose 2014, 21, 3469–3485. [Google Scholar] [CrossRef]
- Pahovnik, D.; Grujić, M.; Cegnar, M.; Kerč, J.; Žagar, E. Synthesis of alkyl-modified poly(sodium glutamate)s for preparation of polymer-protein nanoparticles in combination with N,N,N-trimethyl chitosan. J. Polym. Sci. Part Polym. Chem. 2014, 52, 2976–2985. [Google Scholar] [CrossRef]
- Habraken, G.J.M.; Peeters, M.; Dietz, C.H.J.T.; Koning, C.E.; Heise, A. How controlled and versatile is N-carboxy anhydride (NCA) polymerization at 0 °C? Effect of temperature on homo-, block- and graft (co)polymerization. Polym. Chem. 2010, 1, 514–524. [Google Scholar] [CrossRef]
- Kunishima, M.; Kawachi, C.; Iwasaki, F.; Terao, K.; Tani, S. Synthesis and characterization of 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride. Tetrahedron Lett. 1999, 40, 5327–5330. [Google Scholar] [CrossRef]
- Feng, S.; Bagia, C.; Mpourmpakis, G. Determination of Proton Affinities and Acidity Constants of Sugars. J. Phys. Chem. A 2013, 117, 5211–5219. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Nagai, H.; Noguchi, M.; Kobayashi, A.; Shoda, S. One-step conversion of unprotected sugars to β-glycosyl azides using 2-chloroimidazolinium salt in aqueous solution. Chem. Commun. 2009, 3378–3379. [Google Scholar]
- Haines, A.H. Relative Reactivities of Hydroxyl Groups in Carbohydrates. Adv. Carbohydr. Chem. Biochem. 1976, 33, 11–109. [Google Scholar]
- Ali, S.P.; Jalsa, N.K. Order of Reactivity of OH/NH Groups of Glucosamine Hydrochloride and N-Acetyl Glucosamine Toward Benzylation Using NaH/BnBr in DMF. J. Carbohydr. Chem. 2014, 33, 185–196. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds PBGlu, PGlu and P(Glu-GlcN) are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perdih, P.; Čebašek, S.; Možir, A.; Žagar, E. Post-Polymerization Modification of Poly(L-glutamic acid) with D-(+)-Glucosamine. Molecules 2014, 19, 19751-19768. https://doi.org/10.3390/molecules191219751
Perdih P, Čebašek S, Možir A, Žagar E. Post-Polymerization Modification of Poly(L-glutamic acid) with D-(+)-Glucosamine. Molecules. 2014; 19(12):19751-19768. https://doi.org/10.3390/molecules191219751
Chicago/Turabian StylePerdih, Peter, Sašo Čebašek, Alenka Možir, and Ema Žagar. 2014. "Post-Polymerization Modification of Poly(L-glutamic acid) with D-(+)-Glucosamine" Molecules 19, no. 12: 19751-19768. https://doi.org/10.3390/molecules191219751