
Worldwide Genetic Structure Elucidates the Eurasian Origin and Invasion Pathways of Dothistroma septosporum, Causal Agent of Dothistroma Needle Blight

 , , , , , , , ,
, , , , , , , ,  ,
,  , ,
, ,  , , , , , , add
Show full author list
, , , , , , add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Fungal Isolation
2.2. Haplotype and Mating-Type Determination
2.3. Genetic Diversity and Differentiation
2.4. Mating-Type Distribution and Sexual Recombination
2.5. Population Structure
2.6. Modeling of Evolutionary History
3. Results
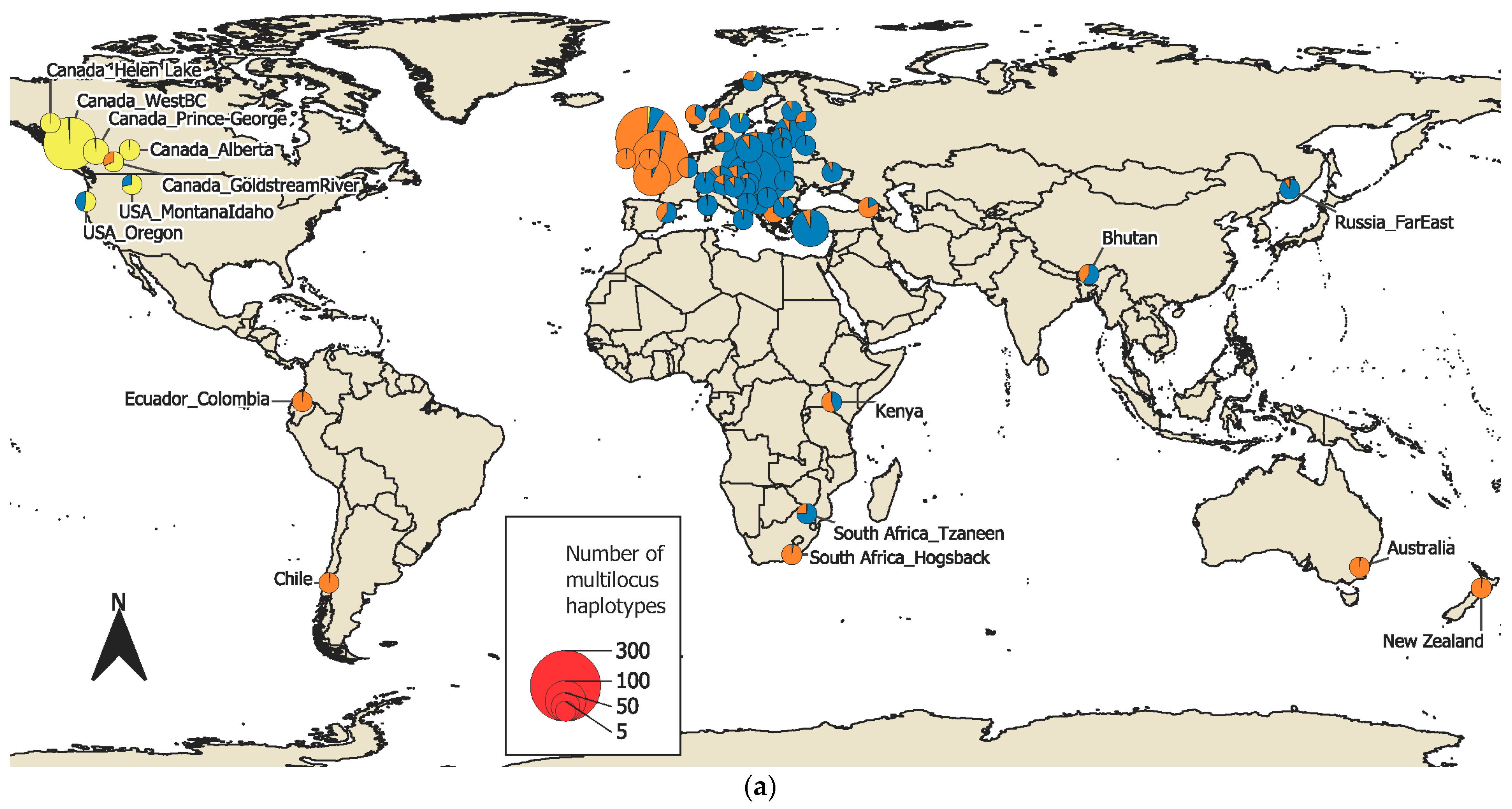
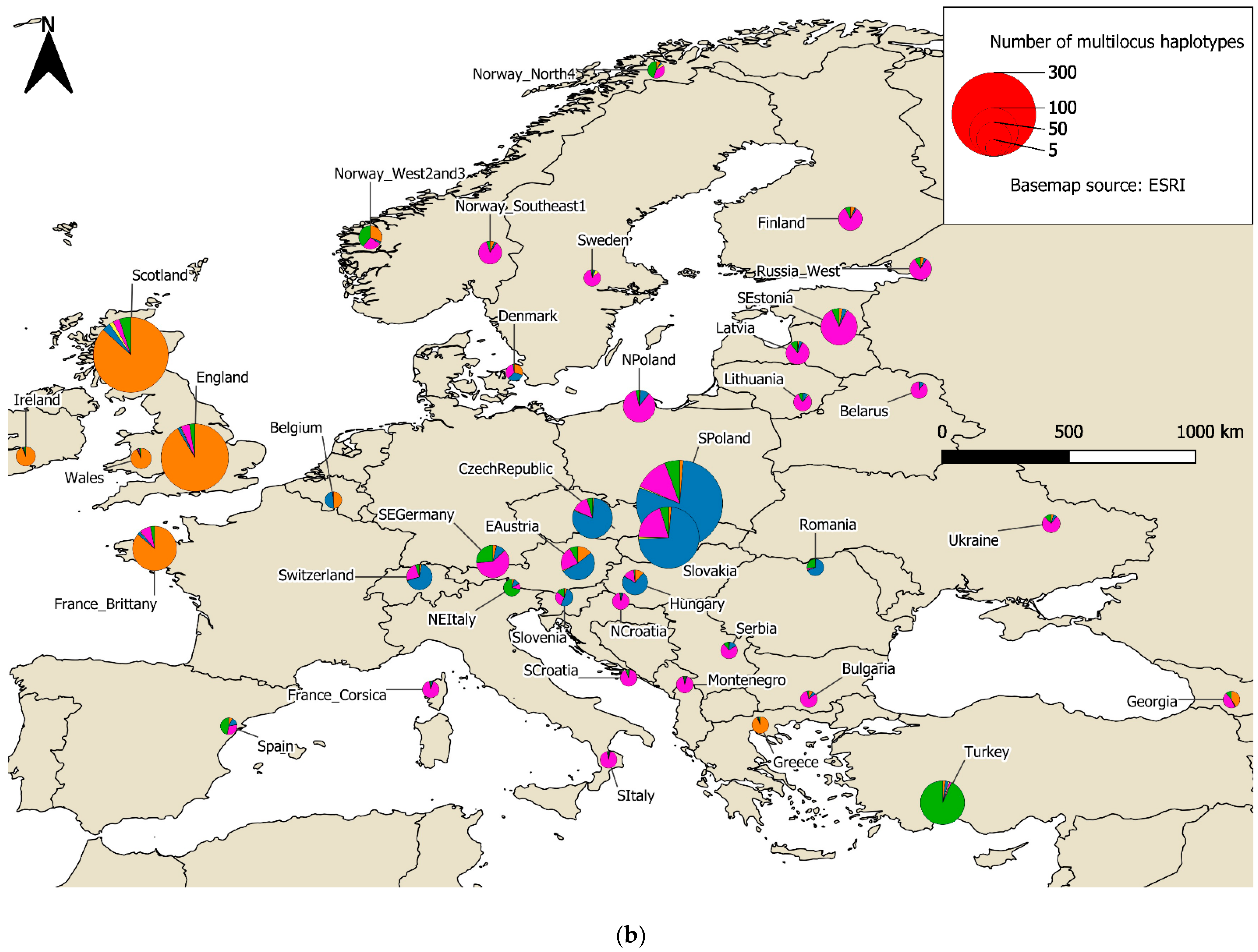
3.1. Isolates and Haplotypes
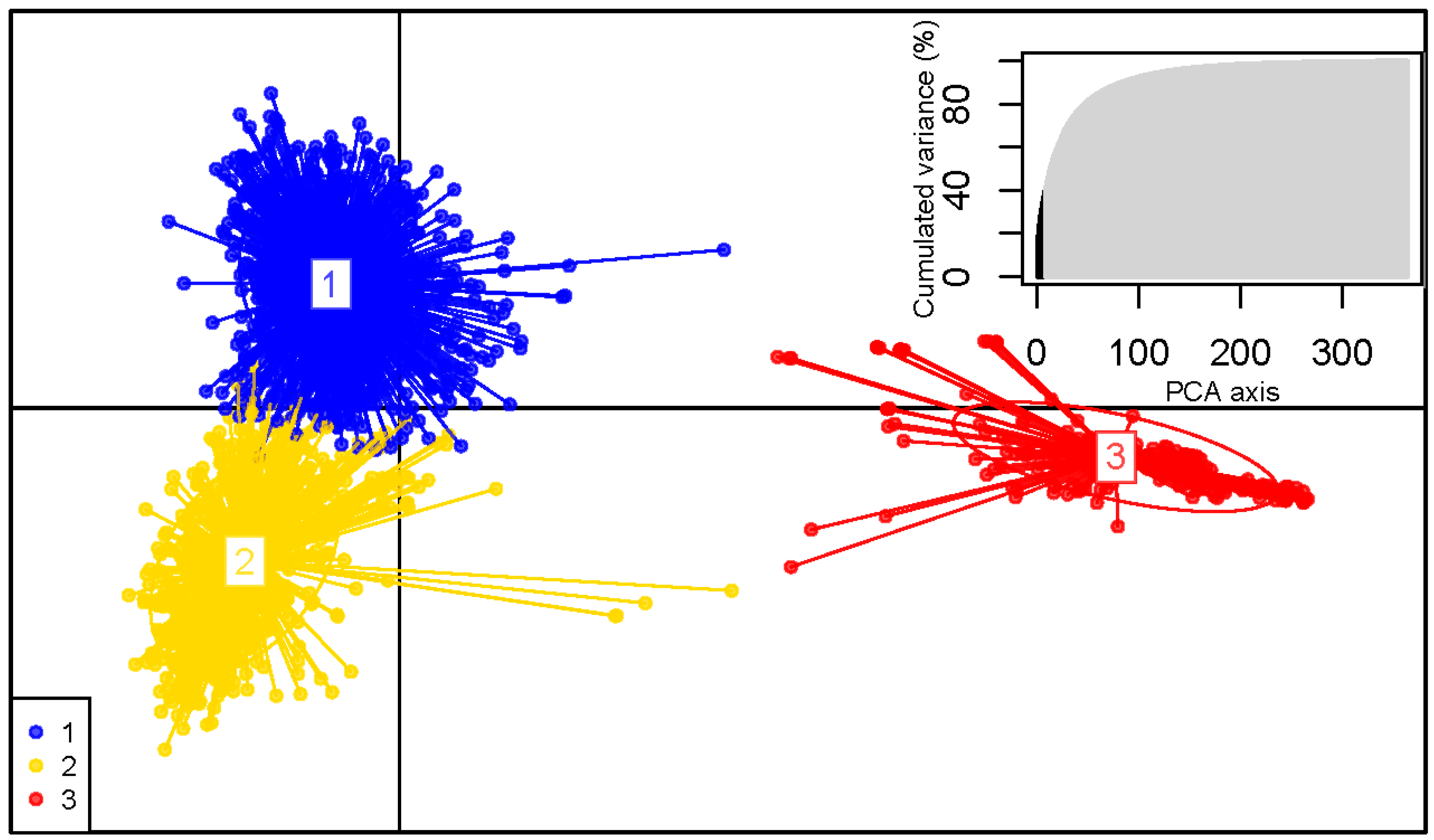
3.2. Population Structure
3.3. Genetic Diversity
3.4. Mating-Type Distribution and Sexual Recombination
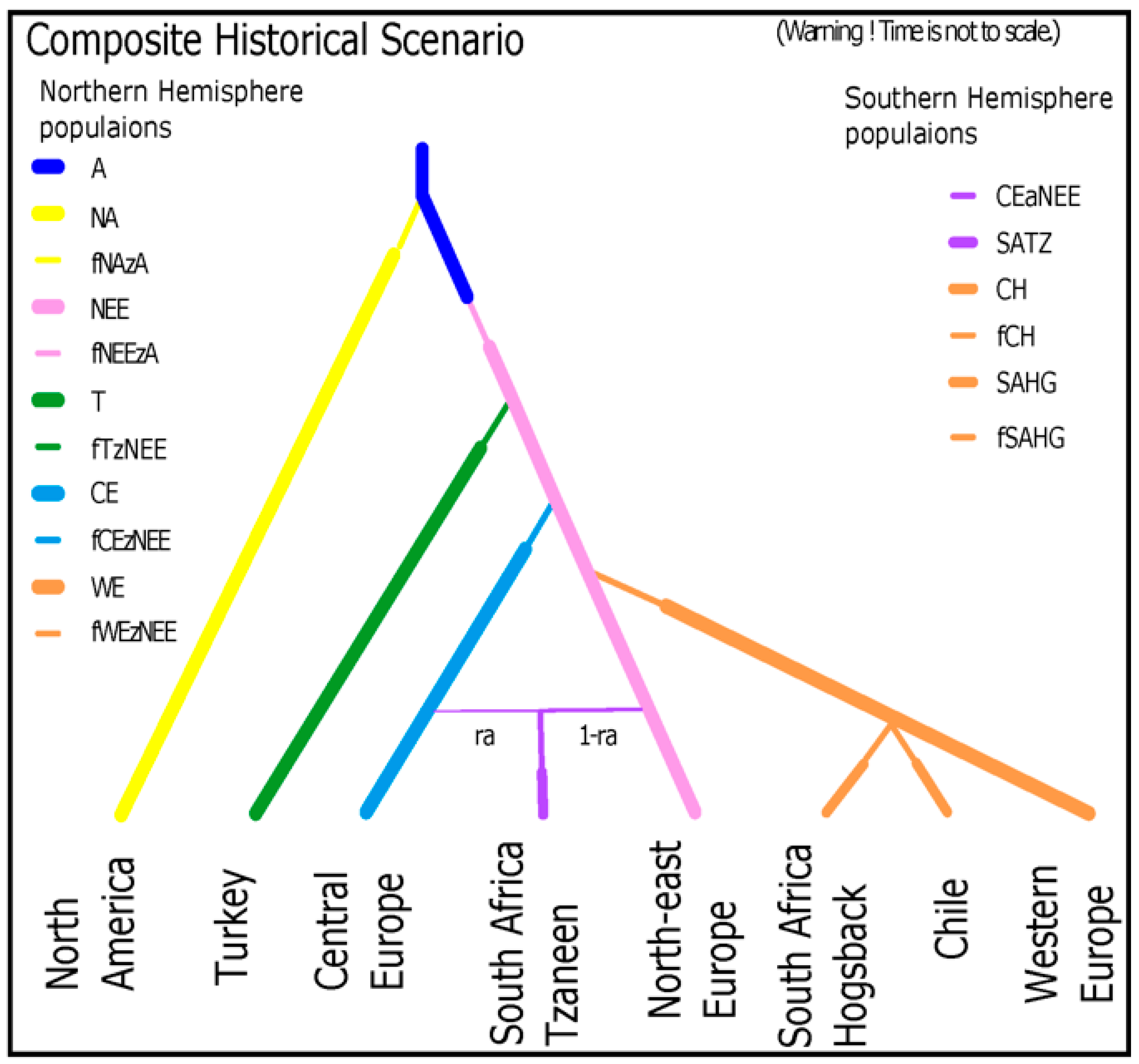
3.5. Modeling of Evolutionary History
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Drenkhan, R.; Tomešová-Haataja, V.; Fraser, S.; Bradshaw, R.E.; Vahalík, P.; Mullett, M.S.; Martín-García, J.; Bulman, L.S.; Wingfield, M.J.; Kirisits, T.; et al. Global Geographic Distribution and Host Range of Dothistroma Species: A Comprehensive Review. For. Pathol. 2016, 46, 408–442. [Google Scholar] [CrossRef]
- Brown, A.V.; Webber, J. Red Band Needle Blight of Conifers in Britain; Forestry Commission: Edinburgh, UK, 2008; p. 8.
- Bulman, L. Foliar Diseases of Pine—The New Zealand Experience. In Proceedings of the 54th Annual Western International Forest Disease Work Conference, Smithers, BC, Canada, 2–6 October 2006; pp. 57–60. [Google Scholar]
- Price, C. Appraising the Economic Impact of Tree Diseases in Britain: Several Shots in the Dark, and Possibly Also in the Wrong Ball-Park? Scand. For. Econ. 2010, 43, 45–61. [Google Scholar]
- Brown, A.V. Dothistroma Needle Blight. Background for Biosecurtiy Workshop 16 February 2011, Forest Research. 2011. Available online: http://saveourwoods.co.uk/wp-content/uploads/2011/03/Tree-Health-Threats-PtLM.pdf (accessed on 28 January 2021).
- Barnes, I.; van der Nest, A.; Mullett, M.S.; Crous, P.W.; Drenkhan, R.; Musolin, D.L.; Wingfield, M.J. Neotypification of Dothistroma septosporum and Epitypification of D. pini, Causal Agents of Dothistroma Needle Blight of Pine. For. Pathol. 2016, 46, 388–407. [Google Scholar] [CrossRef]
- Mullett, M.S.; Adamson, K.; Bragança, H.; Bulgakov, T.S.; Georgieva, M.; Henriques, J.; Jürisoo, L.; Laas, M.; Drenkhan, R. New Country and Regional Records of the Pine Needle Blight Pathogens Lecanosticta acicola, Dothistroma septosporum and Dothistroma pini. For. Pathol. 2018, 48, e12440. [Google Scholar] [CrossRef]
- Gibson, I.A.S. Dothistroma Blight of Pinus radiata. Annu. Rev. Phytopathol. 1972, 10, 51–72. [Google Scholar] [CrossRef]
- Rodas, C.A.; Wingfield, M.J.; Granados, G.M.; Barnes, I. Dothistroma Needle Blight: An Emerging Epidemic Caused by Dothistroma septosporum in Colombia. Plant Pathol. 2016, 65, 53–63. [Google Scholar] [CrossRef]
- Welsh, C.; Lewis, K.J.; Woods, A.J. Regional Outbreak Dynamics of Dothistroma Needle Blight Linked to Weather Patterns in British Columbia, Canada. Can. J. For. Res. 2014, 44, 212–219. [Google Scholar] [CrossRef]
- Woods, A.; Coates, K.D.; Hamann, A. Is an Unprecedented Dothistroma Needle Blight Epidemic Related to Climate Change? Bioscience 2005, 55, 761–769. [Google Scholar] [CrossRef] [Green Version]
- Fabre, B.; Ioos, R.; Piou, D.; Marçais, B. Is the Emergence of Dothistroma Needle Blight of Pine in France Caused by the Cryptic Species Dothistroma pini? Phytopathology 2012, 102, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Doroguine, G. Une Maladie Cryptogamique Du Pin. Bull. Trimest. Société Mycol. Fr. 1911, 27, 105–106. [Google Scholar]
- Evans, H.C. The Genus Mycosphaerella and Its Anamorphs Cercoseptoria, Dothistroma and Lecanosticta on Pines. 1984; p. 102. Available online: https://www.gov.uk/research-for-development-outputs/the-genus-mycosphaerella-and-its-anamorphs-cercoseptoria-dothistroma-and-lecanosticta-on-pines (accessed on 15 January 2021).
- Saccardo, P.A. Mycetes Boreali-Americani. Nuovo G. Bot. Ital. 1920, 27, 75–88. [Google Scholar]
- Thyr, B.D.; Shaw, C.G. Identity of the Fungus Causing Red Band Disease on Pines. Mycologia 1964, 56, 103–109. [Google Scholar] [CrossRef]
- Welsh, C.; Lewis, K.; Woods, A. The Outbreak History of Dothistroma Needle Blight: An Emerging Forest Disease in Northwestern British Columbia, Canada. Can. J. For. Res. 2009, 39, 2505–2519. [Google Scholar] [CrossRef]
- Ivory, M.H. Records of Foliage Pathogens of Pinus Species in Tropical Countries. Plant Pathol. 1994, 43, 511–518. [Google Scholar] [CrossRef]
- Gibson, I.A.S. Impact and Control of Dothistroma Blight of Pines. Eur. J. For. Pathol. 1974, 4, 89–100. [Google Scholar] [CrossRef]
- Adamson, K.; Mullett, M.S.; Solheim, H.; Barnes, I.; Müller, M.M.; Hantula, J.; Vuorinen, M.; Kačergius, A.; Markovskaja, S.; Musolin, D.L.; et al. Looking for Relationships between the Populations of Dothistroma septosporum in Northern Europe and Asia. Fungal Genet. Biol. 2018, 110, 15–25. [Google Scholar] [CrossRef]
- Barnes, I.; Wingfield, M.J.; Carbone, I.; Kirisits, T.; Wingfield, B. Population Structure and Diversity of an Invasive Pine Needle Pathogen Reflects Anthropogenic Activity. Ecol. Evol. 2014, 4, 3642–3661. [Google Scholar] [CrossRef]
- Dale, A.L.; Lewis, K.J.; Murray, B.W. Sexual Reproduction and Gene Flow in the Pine Pathogen Dothistroma septosporum in British Columbia. Phytopathology 2011, 101, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Mullett, M.S.; Brown, A.V.; Fraser, S.; Baden, R.; Tubby, K.V. Insights into the Pathways of Spread and Potential Origins of Dothistroma septosporum in Britain. Fungal Ecol. 2017, 26, 85–98. [Google Scholar] [CrossRef]
- Boroń, P.; Lenart-Boroń, A.; Mullett, M.; Kraj, W.; Grad, B.; Kowalski, T. Temporal Changes in the Population Structure of Dothistroma septosporum at the Site of the First Recorded Outbreak in Poland. Plant Pathol. 2019, 68, 383–391. [Google Scholar] [CrossRef]
- Capron, A.; Feau, N.; Heinzelmann, R.; Barnes, I.; Benowicz, A.; Bradshaw, R.E.; Dale, A.; Lewis, K.J.; Owen, T.J.; Reich, R.; et al. Signatures of Post-Glacial Genetic Isolation and Human-Driven Migration in the Dothistroma Needle Blight Pathogen in Western Canada. Phytopathology 2020. [Google Scholar] [CrossRef] [PubMed]
- Drenkhan, R.; Hantula, J.; Vuorinen, M.; Jankovský, L.; Müller, M.M. Genetic Diversity of Dothistroma septosporum in Estonia, Finland and Czech Republic. Eur. J. Plant Pathol. 2013, 136, 71–85. [Google Scholar] [CrossRef]
- Mullett, M.S.; Brown, A.V.; Barnes, I. Population Structure and Reproductive Mode of Dothistroma septosporum in the Brittany Peninsula of France. Eur. J. Plant Pathol. 2015, 143, 261–275. [Google Scholar] [CrossRef]
- Oskay, F.; Tunalı, Z.; Lehtijärvi, A.T.; Doğmuş-Lehtijärvi, H.T.; Woodward, S.; Mullett, M. Distribution and Genetic Diversity of Dothistroma septosporum in Pinus Brutia Forests of South-western Turkey. Plant Pathol. 2020, 69, 1551–1564. [Google Scholar] [CrossRef]
- Tomšovský, M.; Tomešová, V.; Palovčíková, D.; Kostovčík, M.; Rohrer, M.; Hanáček, P.; Jankovský, L. The Gene Flow and Mode of Reproduction of Dothistroma septosporum in the Czech Republic. Plant Pathol. 2013, 62, 59–68. [Google Scholar] [CrossRef]
- Janoušek, J.; Wingfield, M.J.; Monsivais, J.G.M.; Jankovský, L.; Stauffer, C.; Konečný, A.; Barnes, I. Genetic Analyses Suggest Separate Introductions of the Pine Pathogen Lecanosticta acicola Into Europe. Phytopathology 2015, 106, 1413–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taerum, S.J.; Hoareau, T.B.; Duong, T.A.; de Beer, Z.W.; Jankowiak, R.; Wingfield, M.J. Putative Origins of the Fungus Leptographium procerum. Fungal Biol. 2017, 121, 82–94. [Google Scholar] [CrossRef]
- Guillemaud, T.; Beaumont, M.A.; Ciosi, M.; Cornuet, J.-M.; Estoup, A. Inferring Introduction Routes of Invasive Species Using Approximate Bayesian Computation on Microsatellite Data. Heredity 2010, 104, 88–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boroń, P.; Lenart-Boroń, A.; Mullett, M. The Distribution of Dothistroma septosporum and Its Mating Types in Poland. For. Pathol. 2016, 46, 489–496. [Google Scholar] [CrossRef]
- Mullett, M.; Barnes, I. Dothistroma Isolation and Molecular Identification Methods. 2012. Available online: https://www.forestresearch.gov.uk/documents/305/DIAROD_052012_Isolation_and_indentification_97fNCCI.pdf (accessed on 28 January 2021).
- Bradshaw, R.E.; Ganley, R.J.; Jones, W.T.; Dyer, P.S. High Levels of Dothistromin Toxin Produced by the Forest Pathogen Dothistroma pini. Mycol. Res. 2000, 104, 325–332. [Google Scholar] [CrossRef]
- Groenewald, M.; Barnes, I.; Bradshaw, R.E.; Brown, A.V.; Dale, A.; Groenewald, J.Z.; Lewis, K.J.; Wingfield, B.D.; Wingfield, M.J.; Crous, P.W. Characterization and Distribution of Mating Type Genes in the Dothistroma Needle Blight Pathogens. Phytopathology 2007, 97, 825–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, I.; Cortinas, M.N.; Wingfield, M.J.; Wingfield, B.D. Microsatellite Markers for the Red Band Needle Blight Pathogen, Dothistroma septosporum. Mol. Ecol. Resour. 2008, 8, 1026–1029. [Google Scholar] [CrossRef] [PubMed]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R Package for Genetic Analysis of Populations with Clonal, Partially Clonal, and/or Sexual Reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. 2013. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 28 January 2021).
- Shannon, C.E.; Weaver, W. The Mathematical Theory of Communication; University of Illinois Press: Urbana, IL, USA, 1949. [Google Scholar]
- Grünwald, N.J.; Goodwin, S.B.; Milgroom, M.G.; Fry, W.E. Analysis of Genotypic Diversity Data for Populations of Microorganisms. Phytopathology 2003, 93, 738–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoddart, J.A.; Taylor, J.F. Genotypic Diversity: Estimation and Prediction in Samples. Genetics 1988, 118, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Simpson, E.H. Measurement of Diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Zhan, J.; Pettway, R.E.; McDonald, B.A. The Global Genetic Structure of the Wheat Pathogen Mycosphaerella graminicola Is Characterized by High Nuclear Diversity, Low Mitochondrial Diversity, Regular Recombination, and Gene Flow. Fungal Genet. Biol. 2003, 38, 286–297. [Google Scholar] [CrossRef]
- Nei, M. Estimation of Average Heterozygosity and Genetic Distance from a Small Number of Individuals. Genetics 1978, 89, 583–590. [Google Scholar]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic Analysis in Excel. Population Genetic Software for Teaching and Research—An Update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [Green Version]
- Szpiech, Z.A.; Jakobsson, M.; Rosenberg, N.A. ADZE: A Rarefaction Approach for Counting Alleles Private to Combinations of Populations. Bioinformatics 2008, 24, 2498–2504. [Google Scholar] [CrossRef] [Green Version]
- Excoffier, L.; Lischer, H.E.L. ARLEQUIN Suite Ver 3.5: A New Series of Programs to Perform Population Genetics Analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.H. Handbook of Biological Statistics, 3rd ed.; Sparky House Publishing: Baltimore, MD, USA, 2014; Available online: http://www.biostathandbook.com/exactgof.html (accessed on 28 January 2021).
- Brown, A.H.D.; Feldman, M.W.; Nevo, E. Multilocus Structure of Natural Populations of Hordeum spontaneum. Genetics 1980, 96, 523–536. [Google Scholar] [PubMed]
- Agapow, P.-M.; Burt, A. Indices of Multilocus Linkage Disequilibrium. Mol. Ecol. Notes 2001, 1, 101–102. [Google Scholar] [CrossRef]
- Milgroom, M.G. Recombination and the Multilocus Structure of Fungal Populations. Annu. Rev. Phytopathol. 1996, 34, 457–477. [Google Scholar] [CrossRef] [PubMed]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of Population Structure Using Multilocus Genotype Data: Linked Loci and Correlated Allele Frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A Program for Identifying Clustering Modes and Packaging Population Structure Inferences across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the Number of Clusters of Individuals Using the Software Structure: A Simulation Study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, N.A. Distruct: A Program for the Graphical Display of Population Structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant Analysis of Principal Components: A New Method for the Analysis of Genetically Structured Populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef] [Green Version]
- Jombart, T. Adegenet: A R Package for the Multivariate Analysis of Genetic Markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jombart, T.; Collins, C. A Tutorial for Discriminant Analysis of Principal Components (DAPC) Using Adegenet 2.0.0. 2015. Available online: https://adegenet.r-forge.r-project.org/files/tutorial-dapc.pdf (accessed on 28 January 2021).
- Cornuet, J.-M.; Pudlo, P.; Veyssier, J.; Dehne-Garcia, A.; Gautier, M.; Leblois, R.; Marin, J.-M.; Estoup, A. DIYABC v2.0: A Software to Make Approximate Bayesian Computation Inferences about Population History Using Single Nucleotide Polymorphism, DNA Sequence and Microsatellite Data. Bioinformatics 2014, 30, 1187–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konečný, A.; Estoup, A.; Duplantier, J.-M.; Bryja, J.; Bâ, K.; Galan, M.; Tatard, C.; Cosson, J.-F. Invasion Genetics of the Introduced Black Rat (Rattus Rattus) in Senegal, West Africa. Mol. Ecol. 2013, 22, 286–300. [Google Scholar] [CrossRef]
- Cornuet, J.-M.; Ravigné, V.; Estoup, A. Inference on Population History and Model Checking Using DNA Sequence and Microsatellite Data with the Software DIYABC (v1.0). BMC Bioinform. 2010, 11, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornuet, J.-M.; Santos, F.; Beaumont, M.A.; Robert, C.P.; Marin, J.-M.; Balding, D.J.; Guillemaud, T.; Estoup, A. Inferring Population History with DIY ABC: A User-Friendly Approach to Approximate Bayesian Computation. Bioinformatics 2008, 24, 2713–2719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, R.A.; Liston, A.; Strauss, S.H. Phylogeny and systematics of Pinus. In Ecology and Biogeography of Pinus; Richardson, D.M., Ed.; Cambridge University Press: Cambridge, UK, 2000; pp. 49–68. ISBN 978-0-521-78910-3. [Google Scholar]
- Richardson, D.M.; Rundel, P.W. Ecology and bio- geography of Pinus: An introduction. In Ecology and Biogeography of Pinus; Richardson, D.M., Ed.; Cambridge University Press: Cambridge, UK, 2000; pp. 3–46. ISBN 978-0-521-78910-3. [Google Scholar]
- Fielding, J.M. The Introduction of Monterey Pine into Australia. Aust. For. 1957, 21, 15–16. [Google Scholar] [CrossRef]
- Le Maitre, D.C. Pines in cultivation: A global view. In Ecology and Biogeography of Pinus; Richardson, D.M., Ed.; Cambridge University Press: Cambridge, UK, 2000; pp. 407–431. ISBN 978-0-521-78910-3. [Google Scholar]
- Wu, H.X.; Eldridge, K.G.; Matheson, A.C.; Powell, M.B.; McRae, T.A.; Butcher, T.B.; Johnson, I.G. Achievements in Forest Tree Improvement in Australia and New Zealand 8. Successful Introduction and Breeding of Radiata Pine in Australia. Aust. For. 2007, 70, 215–225. [Google Scholar] [CrossRef]
- de Villebonne, D.; Maugard, F. Rapid Development of Dothistroma Needle Blight (Scirrhia pini) on Corsican Pine (Pinus nigra subsp. laricio) in France. Sante For. Annu. Rep. 1998, 1, 30–32. [Google Scholar]
- Case, T.J.; Taper, M.L. Interspecific Competition, Environmental Gradients, Gene Flow, and the Coevolution of Species’ Borders. Am. Nat. 2000, 155, 583–605. [Google Scholar] [CrossRef]
- Hallatschek, O.; Hersen, P.; Ramanathan, S.; Nelson, D.R. Genetic Drift at Expanding Frontiers Promotes Gene Segregation. Proc. Natl. Acad. Sci. USA 2007, 104, 19926–19930. [Google Scholar] [CrossRef] [Green Version]
- Sexton, J.P.; McIntyre, P.J.; Angert, A.L.; Rice, K.J. Evolution and Ecology of Species Range Limits. Annu. Rev. Ecol. Evol. Syst. 2009, 40, 415–436. [Google Scholar] [CrossRef] [Green Version]
- Rubal, M.; Veiga, P.; Maldonado, C.; Torres, C.; Moreira, J. Population Attributes and Traits of Siphonaria pectinata (Mollusca: Siphonariidae) in Range-Edge and Non Range-Edge Populations at Its Eastern Atlantic Northern Distribution Boundary. J. Exp. Mar. Biol. Ecol. 2015, 471, 41–47. [Google Scholar] [CrossRef]
- Hargreaves, A.L.; Eckert, C.G. Local Adaptation Primes Cold-Edge Populations for Range Expansion but Not Warming-Induced Range Shifts. Ecol. Lett. 2019, 22, 78–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hereford, J. A Quantitative Survey of Local Adaptation and Fitness Trade-Offs. Am. Nat. 2009, 173, 579–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallen, R.M.; Perlin, M.H. An Overview of the Function and Maintenance of Sexual Reproduction in Dikaryotic Fungi. Front. Microbiol. 2018, 9, 503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurst, L.D.; Peck, J.R. Recent Advances in Understanding of the Evolution and Maintenance of Sex. Trends Ecol. Evol. 1996, 11, 46–52. [Google Scholar] [CrossRef]
- Taylor, J.; Jacobson, D.; Fisher, M. The Evolution of Asexual Fungi: Reproduction, Speciation and Classification. Annu. Rev. Phytopathol. 1999, 37, 197–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ennos, R.A.; Sjökvist, E.I.; Piotrowska, M.J.; Riddell, C.; Hoebe, P.N. Using Genome Resequencing to Investigate Racial Structure, Genetic Diversity, Sexual Reproduction and Hybridisation in the Pine Pathogen Dothistroma septosporum. Fungal Ecol. 2020, 45, 100921. [Google Scholar] [CrossRef]
- Piotrowska, M.J.; Riddell, C.; Hoebe, P.N.; Ennos, R.A. Planting Exotic Relatives Has Increased the Threat Posed by Dothistroma septosporum to the Caledonian Pine Populations of Scotland. Evol. Appl. 2018, 11, 350–363. [Google Scholar] [CrossRef]
- Markovskaja, S.; Raitelaitytė, K.; Kačergius, A.; Kolmakov, P.; Vasilevich, V. Occurrence of Dothistroma Needle Blight in Lithuania and Belarus: The Risk Posed to Native Scots Pine Forests. For. Pathol. 2020, e12626. [Google Scholar] [CrossRef]
- Ennos, R.A. The Introduction of Lodgepole Pine as a Major Forest Crop in Sweden: Implications for Host–Pathogen Evolution. For. Ecol. Manag. 2001, 141, 85–96. [Google Scholar] [CrossRef]
- Harrington, T.C.; Wingfield, M.J. Diseases and the ecology of indigenous and exotic pines. In Ecology and Biogeography of Pinus; Cambridge University Press: Cambridge, UK, 1998; pp. 381–401. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
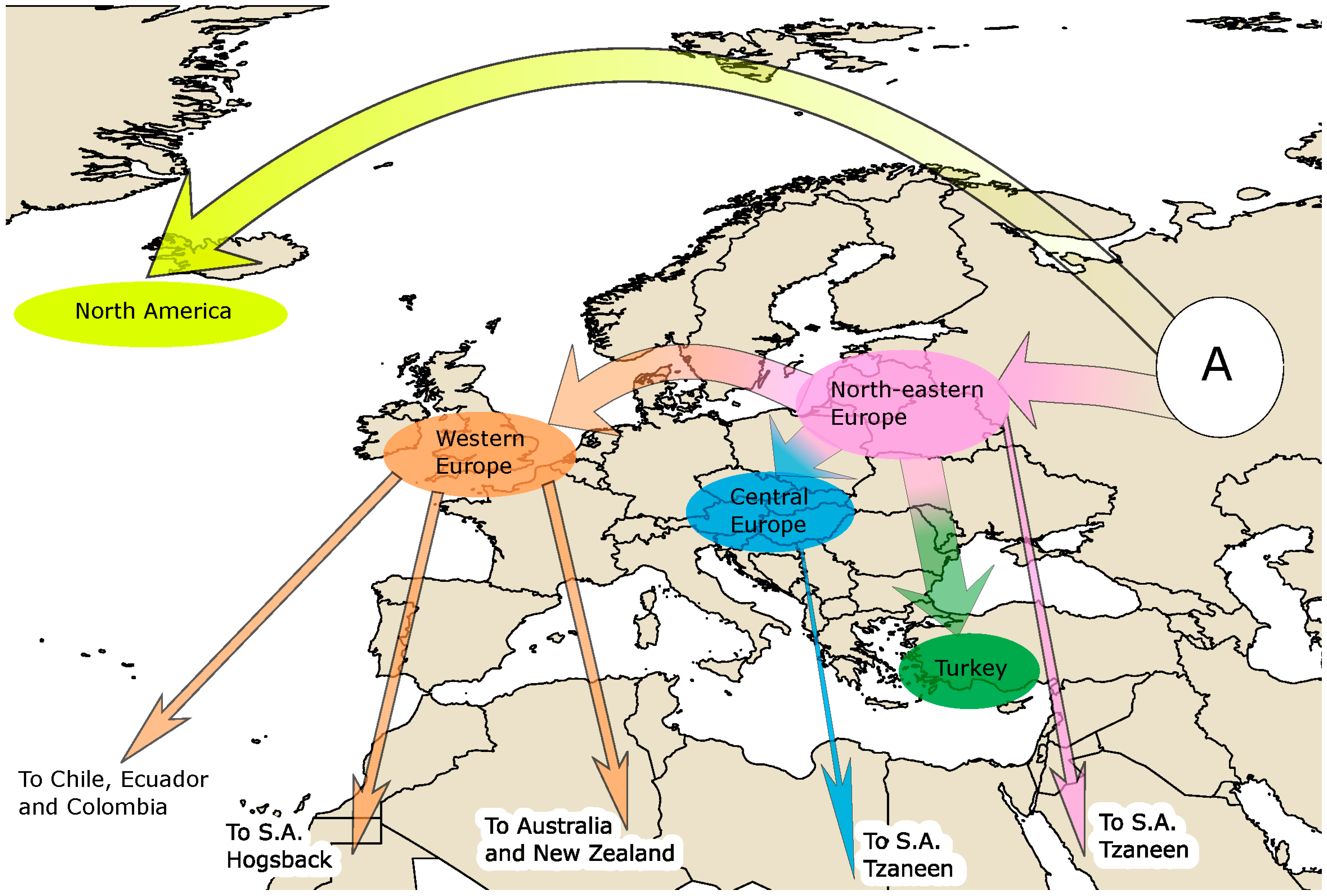{kind=link}
| (a) | ||||||||||||||
| Population Group | N | MLH | eMLH ± Standard Error | H | G | λ | E5 | Hexp | Clonal Fraction | Total Alleles | Private Alleles | h ± Standard Error | AR ± Standard Error | PAR ± Standard Error |
| Australia | 4 | 2 | NA | 0.562 | 1.6 | 0.375 | 0.795 | 0.091 | 0.5 | 12 | 0 | 0.045 ± 0.045 | NA | NA |
| Belarus | 1 | 1 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Belgium | 2 | 2 | NA | 0.693 | 2 | 0.5 | 1 | 0.727 | 0 | 19 | 0 | 0.364 ± 0.07 | NA | NA |
| Bhutan | 12 | 11 | 9.32 ± 0.466 | 2.369 | 10.29 | 0.903 | 0.958 | 0.453 | 0.083 | 37 | 1 | 0.407 ± 0.096 | 2.357 ± 0.353 | 0.208 ± 0.112 |
| Bulgaria | 8 | 8 | NA | 2.079 | 8 | 0.875 | 1 | 0.568 | 0 | 34 | 1 | 0.497 ± 0.069 | 2.531 ± 0.265 | 0.162 ± 0.098 |
| Canada Alberta | 27 | 26 | 9.87 ± 0.334 | 3.244 | 25.14 | 0.96 | 0.979 | 0.353 | 0.037 | 53 | 14 | 0.34 ± 0.108 | 2.11 ± 0.41 | 0.757 ± 0.392 |
| Canada Goldstream River | 1 | 1 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Canada Helen Lake | 4 | 4 | NA | 1.386 | 4 | 0.75 | 1 | 0.394 | 0 | 22 | 0 | 0.295 ± 0.094 | NA | NA |
| Canada Prince-George | 55 | 41 | 9.38 ± 0.736 | 3.585 | 30.56 | 0.967 | 0.843 | 0.518 | 0.255 | 70 | 6 | 0.506 ± 0.077 | 2.46 ± 0.325 | 0.493 ± 0.282 |
| Canada West BC | 239 | 168 | 9.81 ± 0.425 | 4.98 | 119.25 | 0.992 | 0.818 | 0.431 | 0.297 | 113 | 26 | 0.428 ± 0.088 | 2.215 ± 0.307 | 0.601 ± 0.239 |
| Chile | 12 | 11 | 9.32 ± 0.466 | 2.369 | 10.29 | 0.903 | 0.958 | 0.506 | 0.083 | 36 | 0 | 0.453 ± 0.08 | 2.407 ± 0.296 | 0.099 ± 0.053 |
| Czech Republic | 91 | 68 | 9.66 ± 0.561 | 4.111 | 53.43 | 0.981 | 0.874 | 0.598 | 0.253 | 94 | 2 | 0.589 ± 0.071 | 2.7 ± 0.278 | 0.146 ± 0.06 |
| Denmark | 43 | 10 | 4.02 ± 1.099 | 1.454 | 2.89 | 0.654 | 0.577 | 0.543 | 0.767 | 30 | 0 | 0.48 ± 0.075 | 2.366 ± 0.259 | 0.015 ± 0.013 |
| Eastern Austria | 51 | 48 | 9.89 ± 0.316 | 3.85 | 45.63 | 0.978 | 0.97 | 0.654 | 0.059 | 94 | 2 | 0.64 ± 0.062 | 2.938 ± 0.258 | 0.119 ± 0.05 |
| Ecuador and Colombia | 13 | 9 | 7.35 ± 0.764 | 2.032 | 6.26 | 0.84 | 0.794 | 0.278 | 0.308 | 26 | 0 | 0.247 ± 0.07 | 1.459 ± 0.145 | 0.062 ± 0.062 |
| England | 596 | 195 | 8.76 ± 1.024 | 4.259 | 28.96 | 0.965 | 0.401 | 0.554 | 0.673 | 96 | 3 | 0.551 ± 0.074 | 2.522 ± 0.236 | 0.076 ± 0.038 |
| Finland | 26 | 24 | 9.63 ± 0.57 | 3.131 | 21.12 | 0.953 | 0.919 | 0.644 | 0.077 | 72 | 2 | 0.617 ± 0.08 | 2.935 ± 0.321 | 0.203 ± 0.071 |
| France Brittany | 282 | 82 | 7.55 ± 1.342 | 3.402 | 10.27 | 0.903 | 0.319 | 0.584 | 0.709 | 72 | 1 | 0.576 ± 0.076 | 2.656 ± 0.251 | 0.129 ± 0.053 |
| France Corsica | 1 | 1 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Georgia | 2 | 1 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Greece | 3 | 3 | NA | 1.099 | 3 | 0.667 | 1 | 0.455 | 0 | 19 | 0 | 0.303 ± 0.075 | NA | NA |
| Hungary | 30 | 27 | 9.62 ± 0.582 | 3.245 | 23.68 | 0.958 | 0.92 | 0.571 | 0.1 | 64 | 0 | 0.549 ± 0.085 | 2.672 ± 0.313 | 0.094 ± 0.049 |
| Ireland | 20 | 16 | 8.72 ± 0.893 | 2.649 | 11.76 | 0.915 | 0.819 | 0.552 | 0.2 | 36 | 2 | 0.515 ± 0.048 | 2.345 ± 0.151 | 0.104 ± 0.055 |
| Kenya | 9 | 9 | NA | 2.197 | 9 | 0.889 | 1 | 0.349 | 0 | 22 | 0 | 0.31 ± 0.072 | 1.779 ± 0.218 | 0.145 ± 0.142 |
| Latvia | 24 | 23 | 9.84 ± 0.369 | 3.12 | 22.15 | 0.955 | 0.977 | 0.704 | 0.042 | 70 | 0 | 0.672 ± 0.074 | 3.177 ± 0.307 | 0.144 ± 0.082 |
| Lithuania | 16 | 14 | 9.09 ± 0.714 | 2.567 | 11.64 | 0.914 | 0.885 | 0.729 | 0.125 | 66 | 1 | 0.674 ± 0.065 | 3.295 ± 0.299 | 0.219 ± 0.092 |
| Montenegro | 2 | 1 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| North Croatia | 16 | 3 | 2.62 ± 0.489 | 0.777 | 1.86 | 0.461 | 0.727 | 0.394 | 0.813 | 18 | 0 | 0.263 ± 0.078 | NA | NA |
| Northeast Italy | 6 | 5 | NA | 1.561 | 4.5 | 0.778 | 0.93 | 0.346 | 0.167 | 21 | 1 | 0.274 ± 0.085 | 1.764 ± 0.254 | 0.109 ± 0.07 |
| New Zealand | 24 | 5 | 3.07 ± 0.865 | 0.873 | 1.71 | 0.417 | 0.512 | 0.164 | 0.792 | 16 | 0 | 0.131 ± 0.07 | 1.455 ± 0.247 | 0.043 ± 0.035 |
| Norway North | 12 | 6 | 5.17 ± 0.665 | 1.35 | 2.67 | 0.625 | 0.583 | 0.591 | 0.5 | 28 | 0 | 0.462 ± 0.075 | 2.014 ± 0.185 | 0.054 ± 0.039 |
| Norway Southeast | 29 | 23 | 9.33 ± 0.699 | 3.08 | 20.51 | 0.951 | 0.939 | 0.626 | 0.207 | 55 | 1 | 0.589 ± 0.083 | 2.855 ± 0.328 | 0.237 ± 0.12 |
| Norway West | 52 | 23 | 7.41 ± 1.185 | 2.704 | 10.65 | 0.906 | 0.692 | 0.613 | 0.558 | 44 | 0 | 0.581 ± 0.053 | 2.623 ± 0.201 | 0.108 ± 0.049 |
| Northern Poland | 96 | 44 | 8.32 ± 1.124 | 3.336 | 17.39 | 0.943 | 0.605 | 0.585 | 0.542 | 72 | 2 | 0.571 ± 0.083 | 2.704 ± 0.284 | 0.226 ± 0.084 |
| Romania | 3 | 3 | NA | 1.099 | 3 | 0.667 | 1 | 0.606 | 0 | 23 | 0 | 0.404 ± 0.084 | NA | NA |
| Russia Far East | 18 | 15 | 8.97 ± 0.785 | 2.63 | 12.46 | 0.92 | 0.89 | 0.521 | 0.167 | 40 | 1 | 0.48 ± 0.068 | 2.396 ± 0.263 | 0.16 ± 0.094 |
| Russia West | 27 | 21 | 8.91 ± 0.896 | 2.917 | 15.51 | 0.936 | 0.83 | 0.695 | 0.222 | 66 | 1 | 0.661 ± 0.064 | 3.083 ± 0.275 | 0.123 ± 0.069 |
| Scotland | 689 | 240 | 8.82 ± 1.048 | 4.502 | 28.22 | 0.965 | 0.305 | 0.56 | 0.652 | 146 | 22 | 0.557 ± 0.061 | 2.505 ± 0.209 | 0.157 ± 0.099 |
| South Croatia | 8 | 2 | NA | 0.377 | 1.28 | 0.219 | 0.612 | 0.091 | 0.75 | 12 | 1 | 0.045 ± 0.045 | NA | NA |
| Southeast Germany | 64 | 46 | 9.22 ± 0.836 | 3.635 | 28.44 | 0.965 | 0.744 | 0.649 | 0.281 | 92 | 2 | 0.634 ± 0.079 | 2.98 ± 0.308 | 0.333 ± 0.125 |
| Serbia | 9 | 9 | 9 ± 0 | 2.197 | 9 | 0.889 | 1 | 0.487 | 0 | 31 | 0 | 0.428 ± 0.084 | 2.331 ± 0.278 | 0.048 ± 0.031 |
| South Estonia | 61 | 57 | 9.9 ± 0.306 | 4.02 | 53.93 | 0.981 | 0.968 | 0.684 | 0.066 | 116 | 6 | 0.672 ± 0.082 | 3.132 ± 0.335 | 0.218 ± 0.108 |
| South Italy | 2 | 1 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Slovakia | 274 | 157 | 9.54 ± 0.669 | 4.725 | 68 | 0.985 | 0.6 | 0.635 | 0.427 | 120 | 6 | 0.631 ± 0.068 | 2.857 ± 0.273 | 0.103 ± 0.041 |
| Slovenia | 16 | 13 | 8.87 ± 0.743 | 2.513 | 11.64 | 0.914 | 0.938 | 0.62 | 0.188 | 52 | 0 | 0.572 ± 0.075 | 2.852 ± 0.316 | 0.17 ± 0.079 |
| South Africa Hogsback | 20 | 16 | 8.72 ± 0.893 | 2.649 | 11.76 | 0.915 | 0.819 | 0.378 | 0.2 | 26 | 0 | 0.354 ± 0.064 | 1.822 ± 0.153 | 0.015 ± 0.011 |
| South Africa Tzaneen | 14 | 13 | 9.51 ± 0.5 | 2.54 | 12.25 | 0.918 | 0.963 | 0.655 | 0.071 | 40 | 0 | 0.605 ± 0.051 | 2.714 ± 0.237 | 0.018 ± 0.012 |
| Spain | 13 | 2 | 2 ± 0.059 | 0.54 | 1.55 | 0.355 | 0.768 | 0.727 | 0.846 | 19 | 0 | 0.364 ± 0.07 | NA | NA |
| Southern Poland | 602 | 317 | 9.7 ± 0.554 | 5.357 | 112.06 | 0.991 | 0.526 | 0.618 | 0.473 | 115 | 1 | 0.616 ± 0.073 | 2.768 ± 0.27 | 0.074 ± 0.029 |
| Sweden | 8 | 4 | NA | 1.074 | 2.29 | 0.562 | 0.668 | 0.727 | 0.5 | 31 | 0 | 0.54 ± 0.071 | NA | NA |
| Switzerland | 68 | 28 | 6.86 ± 1.333 | 2.659 | 7.39 | 0.865 | 0.481 | 0.58 | 0.588 | 64 | 2 | 0.559 ± 0.068 | 2.665 ± 0.244 | 0.245 ± 0.085 |
| Turkey | 108 | 82 | 9.41 ± 0.79 | 4.181 | 39.14 | 0.974 | 0.592 | 0.625 | 0.241 | 106 | 9 | 0.617 ± 0.083 | 2.871 ± 0.325 | 0.346 ± 0.093 |
| Ukraine | 14 | 14 | 10 ± NaN | 2.639 | 14 | 0.929 | 1 | 0.622 | 0 | 48 | 0 | 0.576 ± 0.052 | 2.713 ± 0.259 | 0.176 ± 0.117 |
| USA Montana and Idaho | 7 | 5 | NA | 1.475 | 3.77 | 0.735 | 0.821 | 0.718 | 0.286 | 35 | 2 | 0.575 ± 0.046 | 3.182 ± 0.263 | 0.489 ± 0.345 |
| USA Oregon | 1 | 1 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Wales | 37 | 18 | 6.46 ± 1.293 | 2.288 | 5.21 | 0.808 | 0.475 | 0.557 | 0.514 | 45 | 0 | 0.525 ± 0.063 | 2.466 ± 0.214 | 0.036 ± 0.02 |
| Total | 3872 | 1913 | 9.85 ± 0.394 | 6.864 | 266.29 | 0.996 | 0.277 | 0.762 | 0.506 | 377 | NA | NA | NA | NA |
| (b) | ||||||||||||||
| Cluster | N | MLH | eMLH ± SE | H | G | λ | E5 | Hexp | Clonal Fraction | Total Alleles | Private Alleles | h ± SE | AR ± Standard Error | PAR ± Standard Error |
| Western European cluster | 1570 | 571 | 190 ± 7.28 | 5.47 | 75 | 0.987 | 0.312 | 0.581 | 0.636 | 152 | 38 | 0.58 ± 0.072 | 11.026 ± 2.809 | 3.150 ± 1.721 |
| Eastern European cluster | 1692 | 1009 | 268 ± 6.6 | 6.54 | 369 | 0.997 | 0.531 | 0.69 | 0.404 | 253 | 112 | 0.69 ± 0.068 | 16.265 ± 3.750 | 6.641 ± 2.524 |
| North American cluster | 331 | 241 | 241 ± 0 | 5.34 | 171 | 0.994 | 0.819 | 0.549 | 0.272 | 157 | 76 | 0.547 ± 0.083 | 14.241 ± 4.045 | 8.175 ± 3.316 |
| Total | 3593 | 1821 | 267 ± 6.96 | 6.9 | 313 | 0.997 | 0.315 | 0.764 | 0.493 | NA | NA | NA | NA | NA |
| (c) | ||||||||||||||
| Eastern European Subcluster | N | MLH | eMLH ± SE | H | G | λ | E5 | Hexp | Clonal Fraction | Total Alleles | Private Alleles | h ± SE | AR ± Standard Error | PAR ± Standard Error |
| Central Europe | 1132 | 653 | 94.8 ± 3.39 | 6.11 | 239.1 | 0.996 | 0.53 | 0.703 | 0.423 | 191 | 44 | 0.634 ± 0.072 | 9.677 ± 2.015 | 1.669 ± 0.595 |
| Northeastern Europe | 303 | 225 | 91.1 ± 3.28 | 5.18 | 111.8 | 0.991 | 0.624 | 0.635 | 0.257 | 174 | 35 | 0.7 ± 0.078 | 11.947 ± 2.904 | 3.557 ± 1.575 |
| Turkey | 108 | 82 | 82 ± 0 | 4.18 | 39.1 | 0.974 | 0.592 | 0.625 | 0.241 | 106 | 21 | 0.617 ± 0.083 | 9.623 ± 1.798 | 3.062 ± 0.68 |
| Total | 1543 | 957 | 98.8 ± 2.96 | 6.52 | 366.9 | 0.997 | 0.538 | 0.692 | 0.38 | NA | NA | NA | NA | NA |
| (a) | ||||||||||||
| Non-Clone-Corrected | Clone-Corrected | |||||||||||
| Population Group | MAT-1-1-1 | MAT 1-2 | p Value | IA | p Value | MAT-1-1-1 | MAT 1-2 | p Value | IA | p Value | ||
| Australia | 0 | 4 | NA | NA | NA | NA | 0 | 2 | NA | NA | NA | NA |
| Belarus | 1 | 0 | NA | NA | NA | NA | 1 | 0 | NA | NA | NA | NA |
| Belgium | 0 | 2 | NA | NA | NA | NA | 0 | 2 | NA | NA | NA | NA |
| Bhutan | 5 | 7 | 0.774 | 0.154 | 0.023 | 0.288 | 5 | 6 | 1 | −0.035 | −0.005 | 0.502 |
| Bulgaria | 2 | 6 | 0.289 | 0.897 | 0.103 | 0.011 | 2 | 6 | 0.289 | 0.897 | 0.103 | 0.009 |
| Canada Alberta | 13 | 14 | 1 | 0.491 | 0.075 | 0.009 | 12 | 14 | 0.845 | 0.43 | 0.067 | 0.014 |
| Canada Goldstream River | 0 | 1 | NA | NA | NA | NA | 0 | 1 | NA | NA | NA | NA |
| Canada Helen Lake | 0 | 4 | NA | NA | NA | NA | 0 | 4 | NA | NA | NA | NA |
| Canada Prince-George | 20 | 35 | 0.058 | 1.67 | 0.172 | <0.001 | 10 | 31 | 0.001 | 1.55 | 0.164 | <0.001 |
| Canada West BC | 123 | 108 | 0.357 | 0.435 | 0.05 | <0.001 | 83 | 77 | 0.693 | 0.28 | 0.032 | <0.001 |
| Chile | 0 | 12 | <0.001 | 0.585 | 0.067 | 0.023 | 0 | 11 | <0.001 | 0.326 | 0.037 | 0.127 |
| Czech Republic | 28 | 60 | <0.001 | 0.194 | 0.02 | 0.002 | 22 | 43 | 0.013 | 0.083 | 0.009 | 0.154 |
| Denmark | 2 | 41 | <0.001 | 4.3 | 0.51 | <0.001 | 1 | 9 | 0.021 | 1.27 | 0.143 | 0.002 |
| Eastern Austria | 22 | 29 | 0.401 | 0.627 | 0.064 | <0.001 | 20 | 28 | 0.312 | 0.59 | 0.06 | <0.001 |
| Ecuador and Colombia | 1 | 12 | 0.003 | 2.32 | 0.337 | <0.001 | 1 | 8 | 0.039 | 2.25 | 0.329 | 0.002 |
| England | 217 | 369 | <0.001 | 0.506 | 0.054 | <0.001 | 85 | 100 | 0.303 | 0.195 | 0.02 | <0.001 |
| Finland | 14 | 12 | 0.845 | 0.82 | 0.085 | <0.001 | 14 | 10 | 0.541 | 0.506 | 0.052 | 0.002 |
| France Brittany | 130 | 151 | 0.233 | 1.98 | 0.213 | <0.001 | 31 | 50 | 0.045 | 0.406 | 0.043 | <0.001 |
| France Corsica | 0 | 1 | NA | NA | NA | NA | 0 | 1 | NA | NA | NA | NA |
| Georgia | 2 | 0 | NA | NA | NA | NA | 1 | 0 | NA | NA | NA | NA |
| Greece | 2 | 1 | NA | NA | NA | NA | 2 | 1 | NA | NA | NA | NA |
| Hungary | 15 | 14 | 1 | 0.615 | 0.065 | 0.002 | 12 | 14 | 0.845 | 0.466 | 0.05 | 0.003 |
| Ireland | 12 | 8 | 0.503 | 0.757 | 0.077 | <0.001 | 8 | 8 | 1.196 | 0.468 | 0.047 | 0.007 |
| Kenya | 6 | 3 | 0.508 | 0.272 | 0.039 | 0.146 | 6 | 3 | 0.508 | 0.272 | 0.039 | 0.147 |
| Latvia | 10 | 14 | 0.541 | 0.45 | 0.046 | 0.32 | 10 | 13 | 0.678 | 0.406 | 0.041 | 0.474 |
| Lithuania | 5 | 10 | 0.302 | 0.399 | 0.042 | 0.06 | 5 | 8 | 0.581 | −0.197 | −0.021 | 0.95 |
| Montenegro | 1 | 1 | NA | NA | NA | NA | 1 | 0 | NA | NA | NA | NA |
| North Croatia | 12 | 4 | 0.077 | 3.26 | 0.664 | <0.001 | 2 | 1 | NA | −0.2 | −0.05 | 0.632 |
| Northeast Italy | 2 | 4 | 0.688 | 0.807 | 0.164 | 0.023 | 2 | 3 | 1 | 0.533 | 0.109 | 0.158 |
| New Zealand | 0 | 24 | <0.001 | 0.134 | 0.071 | 0.224 | 0 | 5 | 0.063 | −0.455 | −0.228 | 0.933 |
| Norway North | 1 | 11 | 0.006 | 4.17 | 0.528 | <0.001 | 1 | 5 | 0.219 | 3.25 | 0.411 | <0.001 |
| Norway Southeast | 20 | 9 | 0.061 | 1.65 | 0.189 | <0.001 | 15 | 8 | 0.21 | 1.45 | 0.165 | <0.001 |
| Norway West | 25 | 27 | 0.89 | 3.55 | 0.356 | <0.001 | 10 | 13 | 0.678 | 2.14 | 0.215 | <0.001 |
| Northern Poland | 56 | 38 | 0.079 | 1.98 | 0.211 | <0.001 | 26 | 16 | 0.164 | 1.33 | 0.139 | <0.001 |
| Romania | 3 | 0 | NA | NA | NA | NA | 3 | 0 | NA | NA | NA | NA |
| Russia Far East | 8 | 10 | 0.815 | 0.645 | 0.072 | 0.054 | 7 | 8 | 1 | 0.477 | 0.053 | 0.236 |
| Russia West | 19 | 7 | 0.029 | 1.69 | 0.171 | <0.001 | 14 | 6 | 0.115 | 0.808 | 0.082 | <0.001 |
| Scotland | 135 | 546 | <0.001 | 1.67 | 0.168 | <0.001 | 78 | 155 | <0.001 | 0.477 | 0.048 | <0.001 |
| South Croatia | 7 | 1 | 0.07 | NA | NA | NA | 1 | 1 | NA | NA | NA | NA |
| Southeastern Germany | 30 | 33 | 0.801 | 0.631 | 0.067 | <0.001 | 21 | 24 | 0.766 | 0.31 | 0.033 | <0.001 |
| Serbia | 5 | 4 | 1 | 0.908 | 0.115 | 0.032 | 5 | 4 | 1 | 0.908 | 0.115 | 0.028 |
| South Estonia | 39 | 20 | 0.018 | 0.466 | 0.05 | 0.129 | 38 | 17 | 0.006 | 0.466 | 0.049 | 0.199 |
| South Italy | 0 | 2 | NA | NA | NA | NA | 0 | 1 | NA | NA | NA | NA |
| Slovakia | 133 | 140 | 0.717 | 0.313 | 0.032 | <0.001 | 77 | 79 | 0.936 | 0.095 | 0.01 | 0.011 |
| Slovenia | 7 | 9 | 0.804 | 0.691 | 0.08 | <0.001 | 6 | 7 | 1 | 0.1 | 0.012 | 0.324 |
| South Africa Hogsback | 8 | 12 | 0.503 | 0.618 | 0.071 | <0.001 | 8 | 8 | 1.196 | 0.386 | 0.044 | 0.009 |
| South Africa Tzaneen | 8 | 6 | 0.791 | 0.903 | 0.092 | <0.001 | 8 | 5 | 0.581 | 0.615 | 0.063 | <0.001 |
| Spain | 13 | 0 | <0.001 | 7 | 1 | <0.001 | 2 | 0 | NA | NA | NA | NA |
| Southern Poland | 255 | 332 | 0.002 | 0.512 | 0.052 | <0.001 | 134 | 172 | 0.034 | 0.327 | 0.033 | <0.001 |
| Sweden | 6 | 2 | 0.289 | 4.72 | 0.526 | <0.001 | 2 | 2 | NA | −0.545 | −0.111 | 0.946 |
| Switzerland | 39 | 25 | 0.103 | 2.77 | 0.287 | <0.001 | 14 | 10 | 0.541 | 1.82 | 0.185 | <0.001 |
| Turkey | 55 | 53 | 0.923 | 0.906 | 0.104 | <0.001 | 45 | 37 | 0.44 | 0.507 | 0.059 | <0.001 |
| Ukraine | 4 | 10 | 0.18 | −0.011 | −0.001 | 0.839 | 4 | 10 | 0.18 | −0.011 | −0.001 | 0.833 |
| USA Montana and Idaho | 6 | 1 | 0.125 | 6.05 | 0.611 | <0.001 | 4 | 1 | 0.375 | 4.5 | 0.514 | <0.001 |
| USA Oregon | 0 | 1 | NA | NA | NA | NA | 0 | 1 | NA | NA | NA | NA |
| Wales | 27 | 9 | 0.004 | 1.58 | 0.165 | <0.001 | 9 | 8 | 1 | −0.027 | −0.003 | 0.544 |
| Total | 1555 | 2259 | <0.001 | 1.02 | 0.104 | <0.001 | 868 | 1057 | <0.001 | 0.969 | 0.099 | <0.001 |
| (b) | ||||||||||||
| Non-Clone-Corrected | Clone-Corrected | |||||||||||
| Cluster | MAT-1-1-1 | MAT 1-2 | p Value | IA | p Value (IAand) | MAT-1-1-1 | MAT 1-2 | p Value | IA | pValue (IAand) | ||
| Western European cluster | 566 | 987 | <0.001 | 0.536 | 0.056 | <0.001 | 220 | 335 | <0.001 | 0.245 | 0.025 | <0.001 |
| Eastern European cluster | 767 | 895 | 0.002 | 0.299 | 0.031 | <0.001 | 459 | 524 | 0.041 | 0.296 | 0.03 | <0.001 |
| North American cluster | 162 | 161 | 1 | 1.193 | 0.125 | <0.001 | 106 | 127 | 0.19 | 1.101 | 0.115 | <0.001 |
| Total | 1554 | 2259 | <0.001 | 1.105 | 0.113 | <0.001 | 828 | 1031 | <0.001 | 1.014 | 0.104 | <0.001 |
| (c) | ||||||||||||
| Non-Clone-Corrected | Clone-Corrected | |||||||||||
| Eastern European Subcluster | MAT-1-1-1 | MAT 1-2 | p Value | IA | p Value (IAand) | MAT-1-1-1 | MAT 1-2 | p Value | IA | pValue (IAand) | ||
| Central Europe | 499 | 609 | 0.001 | 0.257 | 0.026 | <0.001 | 279 | 354 | 0.003 | 0.325 | 0.033 | 0.141 |
| Northeastern Europe | 175 | 122 | 0.002 | 0.498 | 0.051 | <0.001 | 129 | 90 | 0.01 | 0.196 | 0.02 | <0.001 |
| Turkey | 55 | 53 | 0.923 | 0.906 | 0.104 | <0.001 | 45 | 37 | 0.44 | 0.507 | 0.059 | <0.001 |
| Total | 729 | 784 | 0.165 | 0.31 | 0.032 | <0.001 | 453 | 481 | 0.377 | 0.298 | 0.03 | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mullett, M.S.; Drenkhan, R.; Adamson, K.; Boroń, P.; Lenart-Boroń, A.; Barnes, I.; Tomšovský, M.; Jánošíková, Z.; Adamčíková, K.; Ondrušková, E.; et al. Worldwide Genetic Structure Elucidates the Eurasian Origin and Invasion Pathways of Dothistroma septosporum, Causal Agent of Dothistroma Needle Blight. J. Fungi 2021, 7, 111. https://doi.org/10.3390/jof7020111
Mullett MS, Drenkhan R, Adamson K, Boroń P, Lenart-Boroń A, Barnes I, Tomšovský M, Jánošíková Z, Adamčíková K, Ondrušková E, et al. Worldwide Genetic Structure Elucidates the Eurasian Origin and Invasion Pathways of Dothistroma septosporum, Causal Agent of Dothistroma Needle Blight. Journal of Fungi. 2021; 7(2):111. https://doi.org/10.3390/jof7020111
Chicago/Turabian StyleMullett, Martin S., Rein Drenkhan, Kalev Adamson, Piotr Boroń, Anna Lenart-Boroń, Irene Barnes, Michal Tomšovský, Zuzana Jánošíková, Katarína Adamčíková, Emília Ondrušková, and et al. 2021. "Worldwide Genetic Structure Elucidates the Eurasian Origin and Invasion Pathways of Dothistroma septosporum, Causal Agent of Dothistroma Needle Blight" Journal of Fungi 7, no. 2: 111. https://doi.org/10.3390/jof7020111