 Tomasz Jagielski1*†
Tomasz Jagielski1*† Paulina Borówka2,3
Paulina Borówka2,3 Zofia Bakuła1
Zofia Bakuła1 Jakub Lach2,4Błażej Marciniak2,4
Jakub Lach2,4Błażej Marciniak2,4 Anna Brzostek5
Anna Brzostek5 Jarosław Dziadek5
Jarosław Dziadek5 Mikołaj Dziurzyński6Lian Pennings7Jakko van Ingen7
Mikołaj Dziurzyński6Lian Pennings7Jakko van Ingen7 Manca Žolnir-Dovč8
Manca Žolnir-Dovč8 Dominik Strapagiel2,4*†
Dominik Strapagiel2,4*†- 1Department of Applied Microbiology, Faculty of Biology, Institute of Microbiology, University of Warsaw, Warsaw, Poland
- 2Biobank Lab, Department of Molecular Biophysics, Faculty of Biology and Environmental Protection, University of Łódź, Łódź, Poland
- 3Department of Anthropology, Faculty of Biology and Environmental Protection, University of Łódź, Łódź, Poland
- 4BBMRI.pl Consortium, Wroclaw, Poland
- 5Institute of Medical Biology, Polish Academy of Sciences, Łódź, Poland
- 6Department of Bacterial Genetics, Faculty of Biology, Institute of Microbiology, University of Warsaw, Warsaw, Poland
- 7Department of Medical Microbiology, Radboud University Medical Center, Nijmegen, Netherlands
- 8Laboratory for Mycobacteria, University Clinic of Respiratory and Allergic Diseases, Golnik, Slovenia
Only very recently, has it been proposed that the hitherto existing Mycobacterium kansasii subtypes (I–VI) should be elevated, each, to a species rank. Consequently, the former M. kansasii subtypes have been denominated as Mycobacterium kansasii (former type I), Mycobacterium persicum (II), Mycobacterium pseudokansasii (III), Mycobacterium innocens (V), and Mycobacterium attenuatum (VI). The present work extends the recently published findings by using a three-pronged computational strategy, based on the alignment fraction-average nucleotide identity, genome-to-genome distance, and core-genome phylogeny, yet essentially independent and much larger sample, and thus delivers a more refined and complete picture of the M. kansasii complex. Furthermore, five canonical taxonomic markers were used, i.e., 16S rRNA, hsp65, rpoB, and tuf genes, as well as the 16S-23S rRNA intergenic spacer region (ITS). The three major methods produced highly concordant results, corroborating the view that each M. kansasii subtype does represent a distinct species. This work not only consolidates the position of five of the currently erected species, but also provides a description of the sixth one, i.e., Mycobacterium ostraviense sp. nov. to replace the former subtype IV. By showing a close genetic relatedness, a monophyletic origin, and overlapping phenotypes, our findings support the recognition of the M. kansasii complex (MKC), accommodating all M. kansasii-derived species and Mycobacterium gastri. None of the most commonly used taxonomic markers was shown to accurately distinguish all the MKC species. Likewise, no species-specific phenotypic characteristics were found allowing for species differentiation within the complex, except the non-photochromogenicity of M. gastri. To distinguish, most reliably, between the MKC species, and between M. kansasii and M. persicum in particular, whole-genome-based approaches should be applied. In the absence of clear differences in the distribution of the virulence-associated region of difference 1 genes among the M. kansasii-derived species, the pathogenic potential of each of these species can only be speculatively assessed based on their prevalence among the clinically relevant population. Large-scale molecular epidemiological studies are needed to provide a better understanding of the clinical significance and pathobiology of the MKC species. The results of the in vitro drug susceptibility profiling emphasize the priority of rifampicin administration in the treatment of MKC-induced infections, while undermining the use of ethambutol, due to a high resistance to this drug.
Introduction
Non-tuberculous mycobacteria (NTM) comprise all species of the Mycobacterium genus, except those aetiologically implicated in tuberculosis (TB) and leprosy, that is members of the M. tuberculosis complex and M. leprae or M. lepromatosis, respectively. More than 180 NTM species have been recognized to date (LPSN database, 2019). This figure, however, will soon need to be revised, since several new species, on average, are continuously being added every year (Tortoli, 2014). With the increasing number of mycobacterial species descriptions, the number of reported infections, essentially due to NTM, is also growing on a global level. Not as much an enlarging spectrum of NTM species, but more a heightened clinical awareness, expanding population of vulnerable hosts, and advancement of diagnostic and surveillance services are responsible for this scenario (Sood and Parrish, 2017). Although many of the newly described NTM species are potentially pathogenic, having been isolated from clinically affected individuals, only less than a third has consistently been associated with significant health disorders in humans.
Mycobacterium kansasii is one of the most virulent and prevalent NTM pathogen in human medicine. It was first described by Buhler and Pollak in 1953 from a series of respiratory samples of patients with a TB-like pulmonary disease (Buhler and Pollak, 1953). The species was originally named a “yellow bacillus” to emphasize its brilliant yellow pigmentation on exposure to light, but amended thereupon to M. tuberculosis luciflavum by Middlebrook (Middlebrook, 1956) and M. luciflavum by Manten (Manten, 1957). The current species name (M. kansasii) was proposed by Hauduroy in 1955 and refers to where its first isolations were performed (Kansas City, USA) (Hauduroy, 1955). Since the early 1960s, M. kansasii infections have been among the very top of all NTM diseases reported worldwide. A remarkable upsurge in the incidence of M. kansasii infections was seen at the turn of 1980s and 1990s, with the burgeoning of the HIV/AIDS epidemic, and persisted until the first antiretroviral therapies became widely available (Horsburgh and Selik, 1989; Witzig et al., 1995; Santin and Alcaide, 2003). Currently, M. kansasii is one of the six most frequently isolated NTM species across the world. The prevalence of this pathogen is exceptionally high in Slovakia, Poland, and the UK, with the isolation rate of 36, 35, and 11%, respectively, compared to a mean isolation rate of 5% in Europe and 4% globally (Hoefsloot et al., 2013). Chronic, fibro-cavitary lung disease, with upper lobe predominance, and with an overall clinical picture mimicking classical TB, is the most common manifestation attributable to M. kansasii (Matveychuk et al., 2012; Moon et al., 2015; Bakuła et al., 2018b). Much rarer are extrapulmonary infections, such as lymphadenitis, skin and soft-tissue infections, and disseminated disease (Liao et al., 2007; Chen et al., 2008; Park et al., 2012; Shaaban et al., 2014). The exact epidemiology of M. kansasii disease is difficult to ascertain because case reporting is not mandatory in most countries and differentiation between isolation (colonization) and infection may be diagnostically challenging. Moreover, the incidence rates are influenced by a combination of demographic and clinical factors, including patient geographical origin and HIV status. Pulmonary M. kansasii infections tend to cluster in specific geographical areas, such as central Europe or metropolitan centers of London, Brasilia, and Johannesburg (Hoefsloot et al., 2013). Strong epidemiological disparities exist in terms of HIV reactivity. The annual rate of M. kansasii infection among HIV-seropositive patients has been reported to be as high as 532 per 100,000 population, whereas in non-HIV infected individuals it has been calculated at 0.06–2.2 per 100,000 population (Marras and Daley, 2004; Ricketts et al., 2014). Not only the incidence rates, but also the sources of M. kansasii infections and routes of transmission are poorly defined. Similar to other NTM, M. kansasii infections are believed to be acquired from environmental exposures rather than by person-to-person transmission, although a case of interfamilial clustering has been described (Ricketts et al., 2014). Contrary to other NTM, M. kansasii has only sporadically been isolated from soil, natural water systems or animals. Instead, the pathogen has often been recovered from municipal tap water, which is considered its major environmental reservoir (Falkinham, 1996; Thomson et al., 2013).
The genetic structure of M. kansasii was first investigated in the early 1990s. A pioneering work by Ross et al. showed the existence of a genetic subspecies of M. kansasii by sequencing of the 5′ end of the 16S rRNA gene (Ross et al., 1992). Subsequent studies involving the amplification of the 16S-23S rRNA spacer region (Abed et al., 1995), PCR-restriction analysis (PRA) of the highly-conserved hsp65 gene (Plikaytis et al., 1992; Telenti et al., 1993) and Southern blot hybridization with the major polymorphic tandem repeat (MPTR) (Hermans et al., 1992) or insertion sequence-like element, IS1652 (Yang et al., 1993) as a probe have confirmed M. kansasii as a genetically heterogeneous species. The genetic variability of M. kansasii was clearly demonstrated in a study of Picardeau et al. who divided the species into five subspecies or (sub-)types, based on the analysis of restriction fragment length polymorphisms (RFLPs) using the MPTR probe, pulsed-field gel electrophoresis (PFGE), amplified fragment length polymorphism (AFLP) analysis, and PRA of the hsp65 gene (Picardeau et al., 1997). The validity of the five M. kansasii subtypes was further corroborated by sequencing of the 16S-23S rRNA gene or internal transcribed spacer (ITS) region (Alcaide et al., 1997). Somewhat later, two novel types (VI and VII) have been described, according to their hsp65 restriction profiles and sequencing results of the 16S rRNA gene and the 16S-23S rRNA spacer (Richter et al., 1999; Taillard et al., 2003). Moreover, M. kansasii isolates with an intermediate type I (I/II) and atypical type II (IIb) have been reported. Whereas, the former had type I-specific sequence of the hsp65 gene and type II-specific sequence of the spacer region, the latter displayed type II-specific spacer sequence and a unique hsp65 gene sequence (Iwamoto and Saito, 2005). The separateness of the M. kansasii subspecies was further supported by polymorphisms at other genetic loci, including the RNA polymerase gene (rpoB) and the translational elongation factor Tu (tuf ), successfully applied for the differentiation between the subspecies I-VI (Kim et al., 2001; Bakuła et al., 2016). Noteworthy, the inter-subspecies differences have been detected at the protein level. For each of the six (I–VI) M. kansasii subspecies, specific matrix-assisted laser desorption-ionization time-of-flight (MALDI TOF) mass spectral profiles have recently been established (Murugaiyan et al., 2018).
Finally, the genetic diversity of M. kansasii exists not only between the subspecies but also between strains of the same subspecies. This has been repeatedly evidenced upon AFLP, PFGE, repetitive unit (rep-)PCR profiling (Alcaide et al., 1997; Iinuma et al., 1997; Picardeau et al., 1997; Gaafar et al., 2003; Zhang et al., 2004; Wu et al., 2009; Thomson et al., 2014; Kwenda et al., 2015; Bakuła et al., 2018a) and more recently, by a newly developed typing method, based on the analysis of variable number of tandem repeat (VNTR) loci (Bakuła et al., 2018a).
Across all genetic studies performed so far on M. kansasii, a controversy has been growing over the taxonomic rank of the genetic variants of the species, best reflected by their terminology, which includes subspecies, subtypes, and genotypes.
The purpose of this study was to resolve the phylogenetic and taxonomic structure of M. kansasii by combining whole genome sequencing with traditional polyphasic classification approaches. The use of a polyphasic strategy, incorporating phylogenetic, biochemical, and chemotaxonomic criteria for resolving the taxonomic identity of mycobacterial species, especially within the NTM group, has been heavily advocated (Saini et al., 2009).
The issue of molecular taxonomy of M. kansasii has been addressed in a very recent work by Tagini et al. (2019), which appeared shortly before the completion of our own draft. The present study extends the recently published findings by using a new independent sample and somewhat different methodology, and thus delivers a more refined and complete picture of the M. kansasii complex.
Materials and Methods
Strains and Culture Conditions
A total of 27 M. kansasii strains were used in the study (Table 1). Included in this number were 21 single-patient, epidemiologically unrelated, clinical isolates and 6 environmental isolates. The strains were originally recovered before 2016 from 8 countries, i.e., the Netherlands (n = 8), Poland (n = 5), the Czech Republic (n = 4), Spain (n = 2), Belgium (n = 1), Germany (n = 3), South Korea (n = 3), and Italy (n = 1). Patients from whom the strains had been collected were classified as having, or not, a pulmonary NTM disease according to the criteria of the American Thoracic Society (ATS) (Griffith et al., 2007). The type strains of M. kansasii (ATCC 12478T), M. gastri (DSM 43505T), M. marinum (DSM 44344T), M. szulgai (DSM 44166T), M. conspicuum (DSM 44136T), M. riyadhense (DSM 45176T). and M. tuberculosis H37Rv were also included in the study.
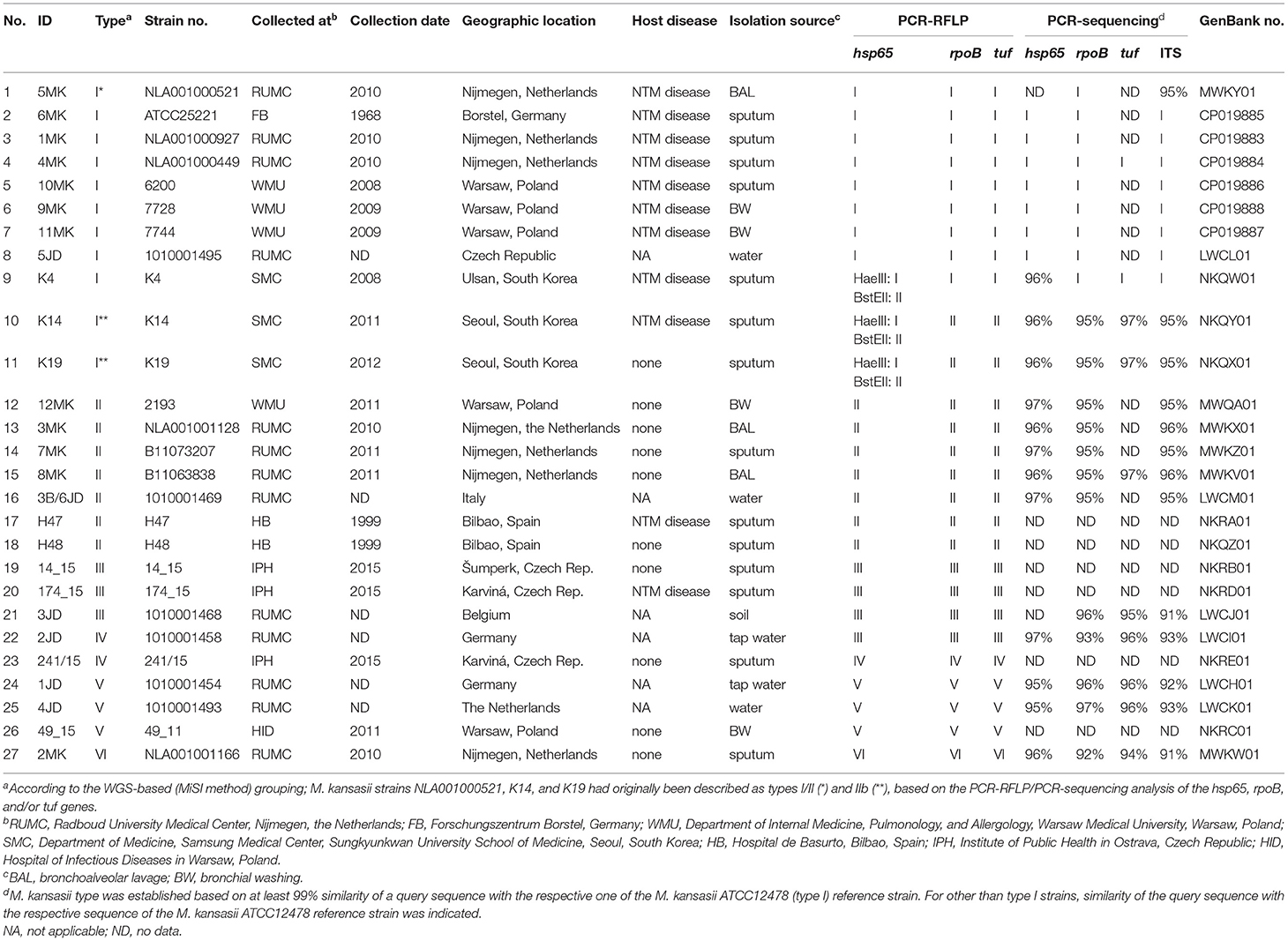
Table 1. Epidemiological, microbiological, and clinical characteristics of M. kansasii strains under the study.
All strains were maintained as frozen stocks and cultured on Löwenstein-Jensen or Middlebrook 7H10 agar (Becton-Dickinson, Franklin Lakes, USA) medium, supplemented with oleic acid, albumin, dextrose, and catalase, and incubated at either 30 or 37°C.
Species Identification and Genotyping
The strains were identified as M. kansasii by using high pressure liquid chromatography (HPLC) of cell wall mycolic acids, in accordance with the Centers for Disease Control and Prevention (CDC) guidelines (Butler et al., 1996) and by the GenoType Mycobacterium CM/AS assay (Hain Lifescience, Nehren, Germany), according to the manufacturer's instructions. For genotypic identification, total DNA was purified from solid bacterial cultures with a standard extraction method described previously (Santos et al., 1992). Genotyping of M. kansasii strains was performed by PCR-RFLP analysis of the hsp65, rpoB, and tuf genes, as reported elsewhere (Telenti et al., 1993; Kim et al., 2001; Bakuła et al., 2016).
Biochemical Profiling
The strains were evaluated for a panel of biochemical characteristics by conventional laboratory procedures (CLSI; Clinical and Laboratory Standards Institute, 2011). These comprised tests for niacin accumulation, nitrate and tellurite reduction, Tween 80 hydrolysis (10 days), catalase (thermostable and semi-quantitative), β-glucosidase, arylsulfatase (3 and 14 days), urease (5 days), and pyrazynamidase. Inhibition tests of tolerance to thiphene-2-carboxylic acid hydrazide (TCH), 5% sodium chloride, and acidic (pH 5.5) conditions were also carried out. In addition, cultural features, including colony morphology, photochromogenicity, the ability to grow on MacConkey agar without crystal violet, and at different temperatures (25, 35, and 45°C) were assessed. For each assay, each strain was tested in triplicate. Only if at least two replications produced identical results, the test was considered complete.
Drug Susceptibility Testing
To test antimicrobial susceptibility, minimal inhibitory concentrations (MICs) for 13 drugs, including isoniazid (INH), rifampicin (RMP), streptomycin (STR), ethambutol (EMB), clarithromycin (CLR), amikacin (AMK), rifabutin (RFB), moxifloxacin (MFX), ciprofloxacin (CFX), co-trimoxazole (SXT), linezolid (LZD), doxycycline (DOX), and ethionamide (ETO), were determined by the microdilution method, using the Sensititre® SLOMYCO plates (TREK Diagnostic Systems, Cleveland, USA), following the Clinical and Laboratory Standards Institute (CLSI) recommendations (CLSI; Clinical and Laboratory Standards Institute, 2011).
Genome Sequencing and Assembly
For the whole-genome sequencing, chromosomal DNA from all 27 M. kansasii strains under the study was extracted by mechanical cell (100 mg pellet) disruption by using zirconia ceramic beads in a FastPrep-24 instrument (MP Biomedicals, Valiant Co., Yantai, China) and further extracted chemically followed with a DNAzol® reagent (Invitrogen, Carlsbad, USA).
DNA concentration, its purity and integrity was determined using a Qubit high-sensitivity (HS) assay kit (ThermoFisher, Waltham, USA).
Paired-end libraries were prepared from 1 ng of high-quality genomic DNA with the Nextera XT DNA sample preparation kit according to the manufacturer's instructions (Illumina Inc., San Diego, USA). The libraries were sequenced on a HiSeq 2500 or a NextSeq 500 instrument (Illumina, San Diego, USA) at a read length of 2 × 150 bp. The quality of reads before and after pre-processing was assessed using FastQC (v0.11.5) (Andrews, 2010). The raw reads were trimmed with TrimGalore ver. 0.43 (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), and de novo assembled with the SPAdes Genome Assembler ver. 3.10.0 (Nurk et al., 2013). Scaffold-level assembly was tested using Quast (QUality ASsesment Tool) (Gurevich et al., 2013) to generate basic genome statistics.
In addition to genomes of 27 M. kansasii strains, sequenced in this study, genomes of other 53 Mycobacterium sp. strains were analyzed. The genomic sequences of these strains were retrieved from the GenBank database (http://www.ncbi.nlm.nih.gov/genbank/) and their appropriate accession numbers were provided in Supplementary Table 6.
Genomic Data Availability
The assembled genomes were deposited under NCBI Bio-Project accession numbers: PRJNA374853 and PRJNA317047. The genomes were deposited in the GenBank under accession numbers provided in Table 1 and Supplementary Table 6.
Annotation and Comparative Genome- and Gene-Scale Analyses
Gene identification and annotation were achieved using the DFAST pipeline v1.0.5 with default settings (Tanizawa et al., 2017).
The genomic relatedness between the strains analyzed was assessed using the MiSI (Microbial Species Identifier) method, based on a combination of genome-wide average nucleotide identity (gANI) and alignment fraction (AF) of orthologous genes (Varghese et al., 2015). The gANI and AF values of 96.5 and 0.6, respectively, were assumed as cutoffs for species delimitation.
As a second method to evaluate the genomic relatedness, the Genome-to-Genome Distance Calculator (GGDC at http://ggdc.dsmz.de) was used. This algorithm was designed to replace standard DNA–DNA hybridization (DDH) by calculating DNA-DNA relatedness (Meier-Kolthoff et al., 2013a). The genome-to-genome distance (GGD) value of 0.0258 was identified as a maximum threshold to assign a genome pair to the same species.
The pairwise ANI and AF values were determined from using ANIcalculator (v1.0) (Yoon et al., 2017).
Full-length sequences of the 16S rRNA (1537 bp; MH794239.1), hsp65 (664 bp; NC_000962.3:528608–530230), rpoB (3439 bp; C_000962.3:759807–763325), tuf (1191 bp; NC_000962.3:784821–786011) genes, and the 16S-23S rRNA intergenic spacer region (ITS) (358 bp; AL123456.3:1473359–1473716) were extracted from the whole-genome sequence of the M. tuberculosis reference strain H37Rv, using CLC Genomic Workbench 10 (Qiagen, Valencia, USA), and were used, each, as a reference for alignments with the respective sequences of other mycobacterial species. Sequences from the same loci of the draft genomes analyzed were identified using the blastn algorithm. Due to a significant variation of the sought sequences, whenever necessary, they were extracted manually or downloaded from the NCBI database (https://www.ncbi.nlm.nih.gov/).
Identification of RD1-14 Genes
To establish the presence of genes within 14 regions of deletions RD1-14, DIFFIND software with -c 0.7 (sequence identity threshold) and -s2 0.5 (length difference cut-off) parameters was used (Marciniak et al., 2017). Identification of RD1-14 genes was based on the alignment of amino acid sequences of predicted genes from the analyzed genomes to the reference database. As references were used amino acid sequences produced by RD1-14 genes from the M. tuberculosis reference strain H37Rv (Accession no.: NC_000962.3) and listed by Brosch et al. (2002).
Phylogenetic Analysis
Core-genome single-ortholog tree was built as described previously (Tagini et al., 2019). Briefly, OrthoFinder (ver. 2.3.4) and MAFFT (ver. 7.310) were used to create core-genome concatemers for the analyzed genomes (Katoh and Standley, 2013; Emms and Kelly, 2019), which were submitted to FastTree (version 2.1.11 Double precision) for maximum-likelihood core-genome phylogeny calculations (Price et al., 2010). The tree was midpoint rooted in FigTree version 1.4.4 (https://github.com/rambaut/figtree).
For single-gene phylogenies, the respective sequences of each genetic locus were subjected to multiple alignment in MEGA X software (ClustalW algorithm) (Kumar et al., 2018). The resulting fragments were further trimmed to remove unnecessary gaps or regions, to a final length of 1,537, 644, 3,439, 1,180, and 277 bp for 16S RNA hsp65, rpoB, tuf, and 16S-23S rRNA ITS region, respectively. The so prepared sequences were used for evolutionary distance calculation according to the Jukes-Cantor model (Jukes and Cantor, 1969). Phylogenetic trees for each target locus sequences were built using the neighbor-joining method and midpoint rooted with the MEGA X software (Saitou and Nei, 1987). Tree topologies were evaluated by bootstrap analysis based on 1,000 replications (Felsenstein, 1985).
Furthermore, a concatenated tree was constructed based on the 16sRNA, rpoB, and hsp65 gene sequences combined. Sequence concatenation was performed with Sequence Matrix 1.8 software (Vaidya et al., 2011), further corrected in Geneious Prime (Kearse et al., 2012), and plotted in MEGA X. Sequence similarity matrices were generated using Bioedit ver. 7.0.5 (IDENTIFY matrice) (Hall, 1999).
Results and Discussion
Sequencing of 27 M kansasii genomes yielded a mean coverage of 66.4x per genome. The average number of contigs per genome was 129 (±229) corresponding to an average N50 score of 4933 kb (±2,605 kb).
The genome sizes and GC contents ranged from 5.6 to 6.6 Mbp (avg. 6.2 Mbp ± 0.3 Mbp) and from 65.86 to 66.38 (avg. 66.14 ± 0.10), respectively. These values were consistent with the data reported for previously assembled M. kansasii genomes (Table 2).
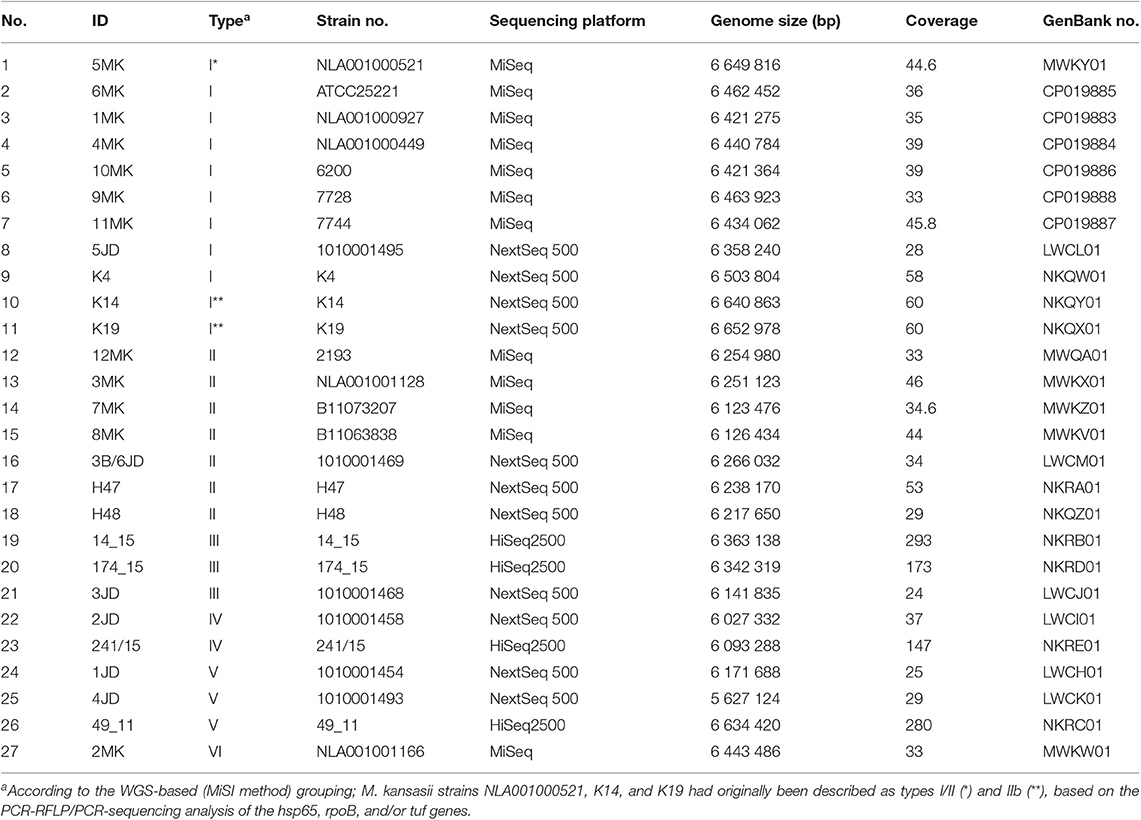
Table 2. Genome features of M. kansasii strains under the study.
The whole-genome-level relatedness among the 27 M. kansasii strains under the study was assessed with three species identification-relevant parameters, namely the alignment fraction (AF) of orthologous genes, the average nucleotide identity (ANI), and the genome-to-genome distance (GGD) (Tables 3 and 4). The AF/ANI metrics were also computed for genomic sequences of another 32 M. kansasii strains and 24 Mycobacterium sp. strains, representing 5 NTM species and 5 M. tuberculosis complex species, all extracted from NCBI databases (Supplementary Table 6). Whereas, the GGD analysis was performed on 69 genomes in total, including 59 M. kansasii genomes and 10 genomes representing single NTM (other than M. kansasii) and M. tuberculosis complex species (Supplementary Table 6).
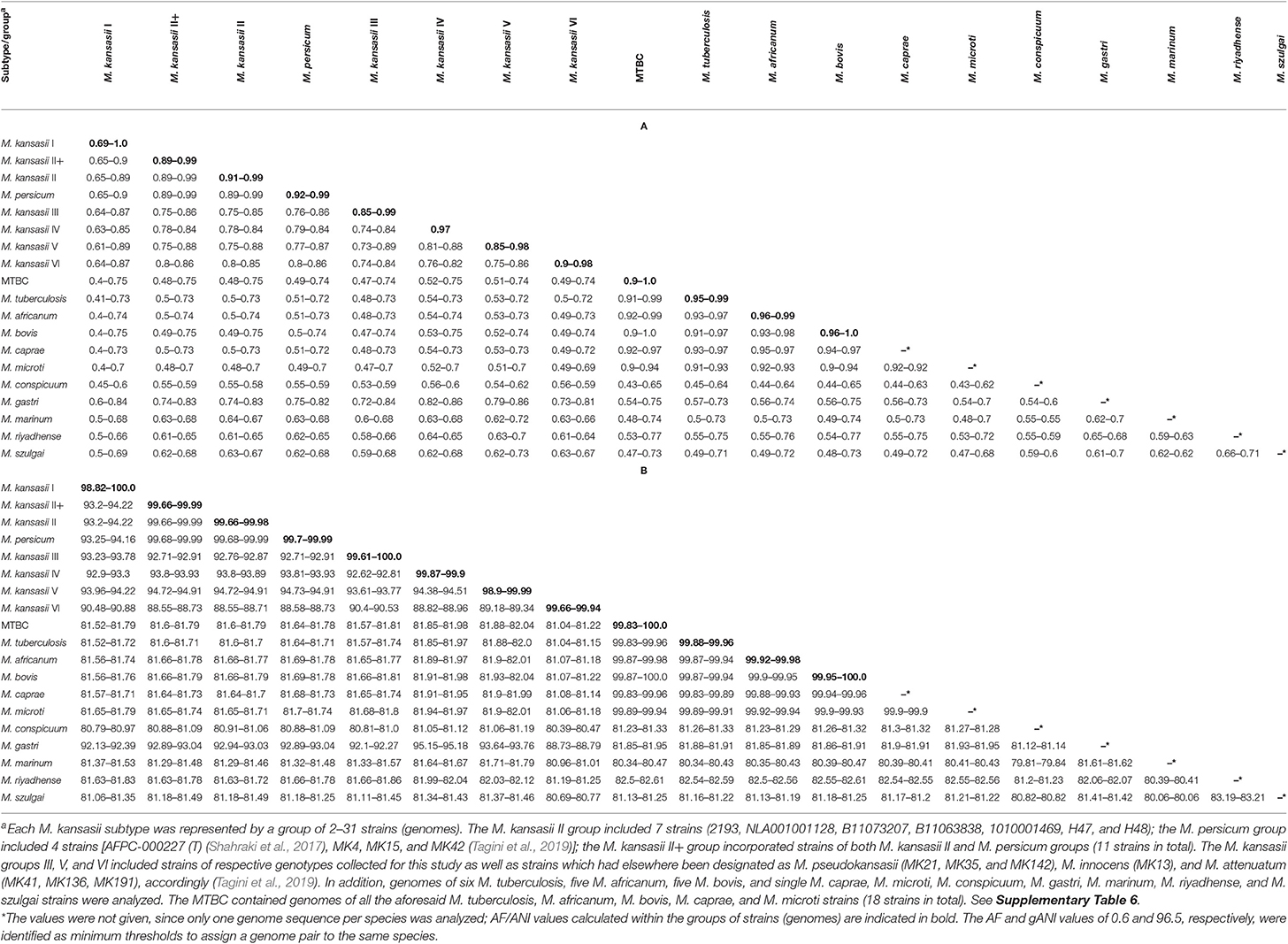
Table 3. Pairwise alignment fractions (A) and average nucleotide identities (B) for M. kansasii subtypes and outgroup species.
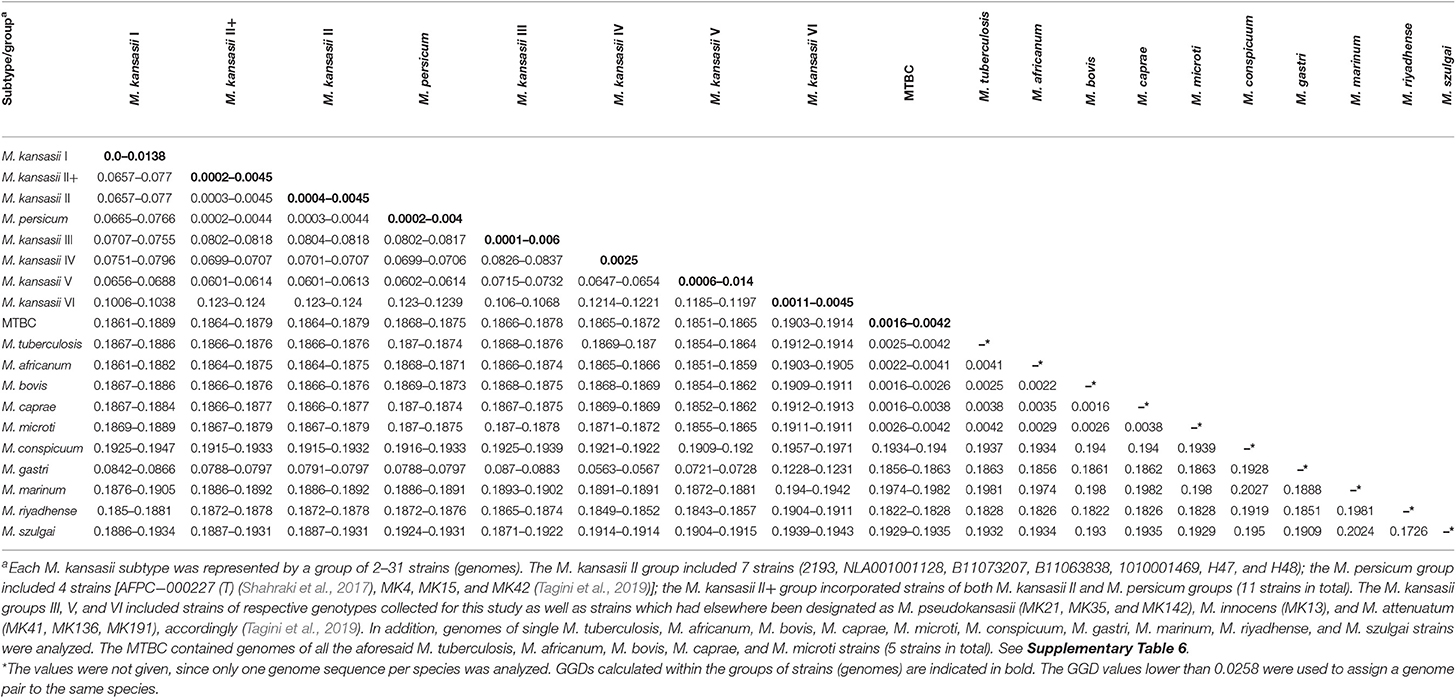
Table 4. Genome-to-genome distance (GGD) of M. kansasii subtypes and other mycobacterial outgroup species.
The AF values for all M. kansasii strains ranged between 0.6 and 1.0. The lowest AF value recorded for two strains, members of the same M. kansasii genotype was 0.69 (type I, range 0.69–1.0). Within all other types, the AF values were 0.85 or higher.
All M. kansasii genotypes showed AF values equal to or below 0.75 with strains of other Mycobacterium species, except for M. gastri and M. persicum, which yielded AF values as high as 0.86 with M. kansasii type IV and V (range, 0.79–0.86) or 0.99 with M. kansasii type II (range, 0.89–0.99), respectively (Table 3A).
Pairwise ANI values for any M. kansasii strains affiliated with different types never exceeded the 95–96% threshold (range, 88.6–94.9%), commonly used as a boundary of species delineation (Richter and Rosselló-Móra, 2009; Varghese et al., 2015). Whereas, the ANI values for strains of the same M. kansasii genotype were always higher than 98.8%. The ANI values between M. kansasii and other Mycobacterium species were all below 95% (range, 80.4–94.9%) except between M. kansasii genotype II and M. persicum (range, 99.7–99.9%) and between M. kansasii genotype IV and M. gastri (95.2%) (Table 3B).
The AF and ANI metrics were also analyzed combinatorially, using the Microbial Species Identifier (MiSI) algorithm. The MiSI method sorts the analyzed genomes into species-like taxa or cliques, based on the AF and ANI species-level cut-off values set at 0.6 and 96.5%, respectively (Varghese et al., 2015). For a total of 83 mycobacterial genomes studied, 15 different cliques were configured. The results were pictorially summarized in Figure 1. The genomes of 59 M. kansasii strains were clearly divided into six cliques, each containing strains of a distinct M. kansasii subtype (I–VI) only. All the remaining NTM species had their genomes clustered within separate cliques, except that four genomes of strains classified as M. persicum were allocated in the M. kansasii genotype II clique. The genomes of 19 strains, representing five M. tuberculosis complex species were accommodated in a single cluster (clique).
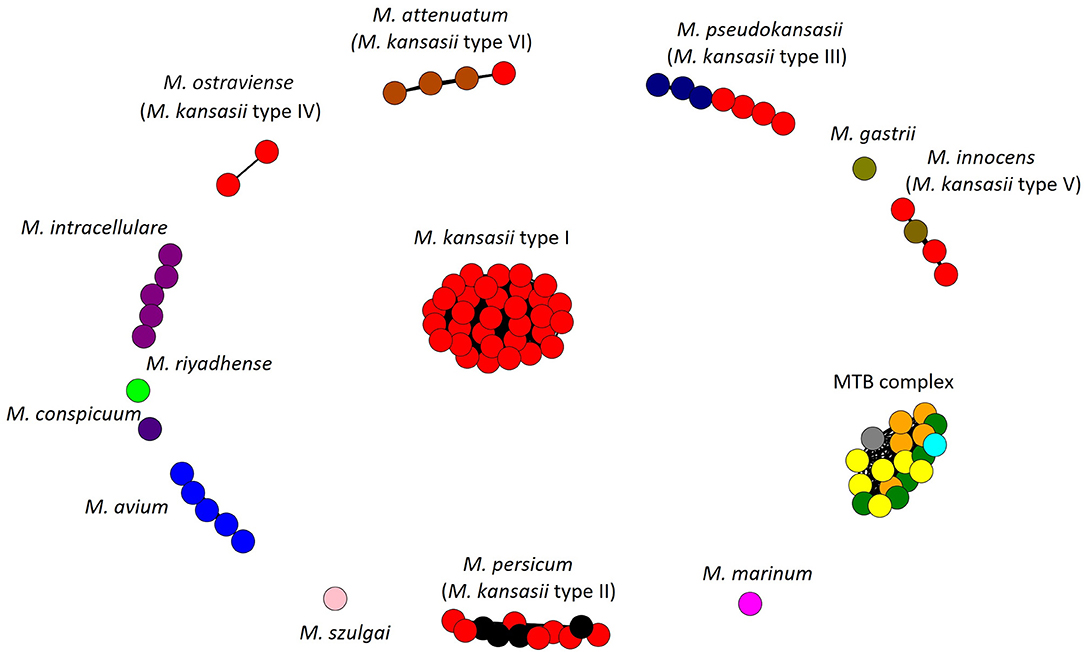
Figure 1. Application of the Microbial Species Identifier (MiSI) method in classifying Mycobacterium kansasii subtype I-VI strains. Separation of different species into different cliques is depicted. Each color represents a species, while each circle—a genome of that species. Within cliques corresponding to M. persicum (formerly subtype II), M. pseudokansasii (III), M. innocens (V), and M. attenuatum (VI), genomes, which had been analyzed previously and first assigned to newly proposed species are shown in other than red color. Within the M. tuberculosis (MTB) complex clique, genomes of M. tuberculosis, M. africanum, M. bovis, M. caprae, and M. microti are depicted in yellow, orange, green, blue, and gray, respectively.
The results of the AF/ANI calculations were fully corroborated by the GGD analysis, which serves as an in silico equivalent of the laboratory-based DNA-DNA hybridization (DDH) (Auch et al., 2010; Meier-Kolthoff et al., 2013b). Here, all strains contained within the M. kansasii genotypes shared enough sequence similarity to be considered as separate species (Table 4). The intra-genotype GGD values ranged from 0.0 to 0.0138, and thus fell under the recommended cut-off value of ≤0.0258, corresponding to a 70% DDH cut-off, for species demarcation (Meier-Kolthoff et al., 2013b). Much higher were the GGD values between strains representing different M. kansasii genotypes (range, 0.0601–0.124) or between any M. kansasii and any other Mycobacterium species (range, 0.0563–0.1971). The only exception was when comparing genomes of M. kansasii type II to any of M. persicum, for which the GGD values were between 0.0003 and 0.0044 (Table 4).
Another whole genome-level approach to clarify the taxonomic relationships between the six M. kansasii subtypes was the core-genome phylogenetic analysis. A dendrogram based on the concatenated 615,565-amino-acid sequences from 1,752 single-copy orthologous genes clearly separated all M. kansasii subtypes (Figure 2). All strains belonging to the same subtype formed distinct clades, supported by high bootstrap values (76.7–100%). Noteworthy, strains of M. persicum located in the same clade as the M. kansasii type II strains, whereas M. gastri branched sisterly to M. kansasii type IV. The topology of the tree supports the separation of M. kansasii subtypes, as distinct species, with M. kansasii type II being conspecific with M. persicum.
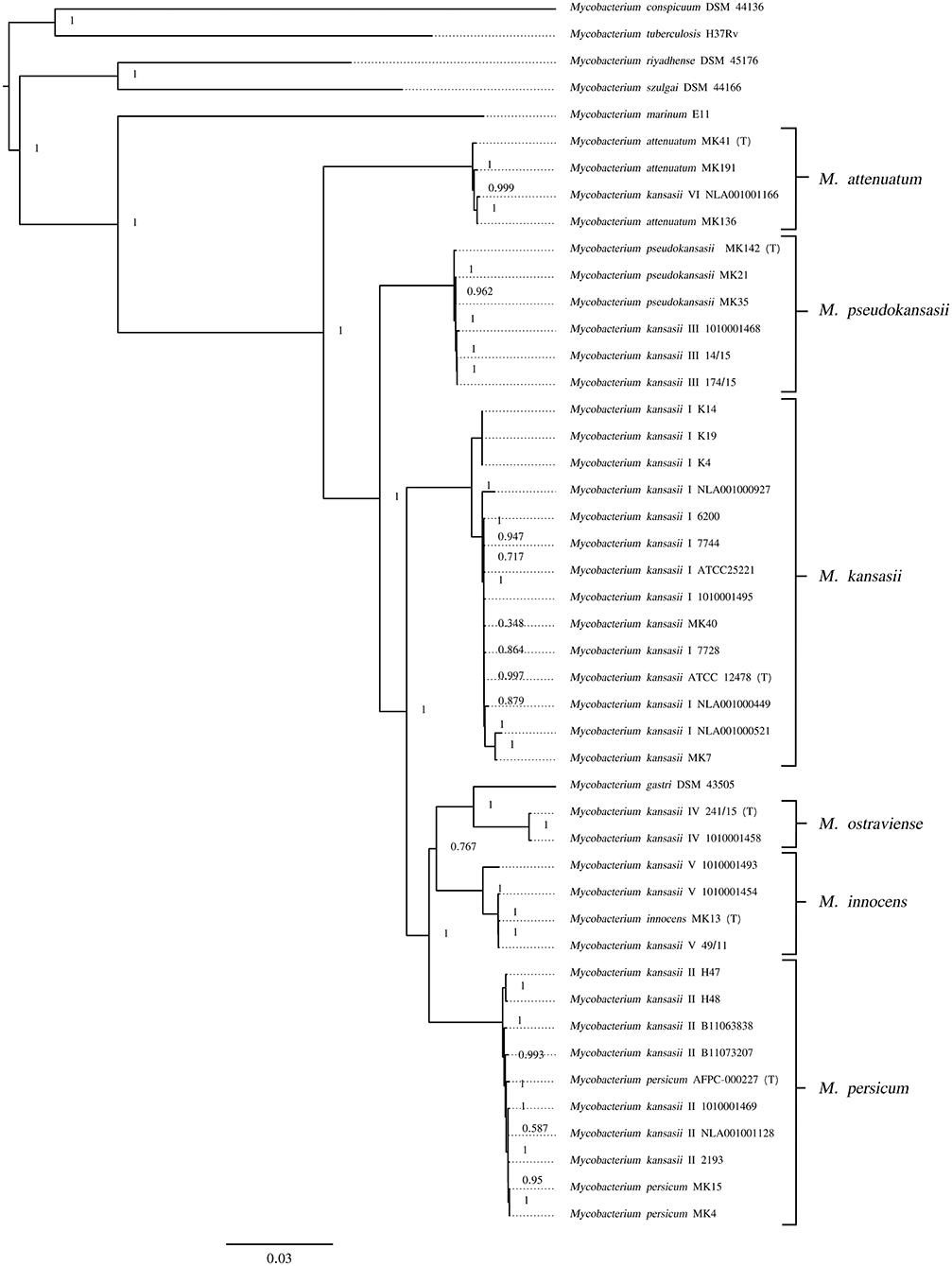
Figure 2. Maximum-likelihood phylogenetic tree based on the amino acid alignment of concatenated single-copy orthologous genes. Bar, number of amino acid substitutions per amino acid site. Node supports were computed using the Shimodaira-Hasegawa test.
The taxonomic position of M. kansasii genotypes was also examined using phylogenetic inferences from five genetically conserved loci, including the canonical 16S rRNA gene, the ITS region, and three protein-coding genes, namely hsp65, tuf, and rpoB, all being widely applied as molecular markers for the classification of mycobacteria (Tortoli, 2014). Multi-alignment and phylogenetic analyses for each of the five loci was conducted on the sequences of all 59 M. kansasii strains and single strains of M. tuberculosis and five other NTM species (Supplementary Table 6).
Pairwise alignments of the 1,537-bp sequences of 16S rRNA gene from M. kansasii strains showed that they were highly similar or identical (99–100% sequence similarity), both within and between the subtypes (Supplementary Table 1). At the same time, strains of M. kansasii shared 98–98.6% similarity with M. tuberculosis and more than 98% similarity with other NTM species. The 16S rRNA gene sequences were identical between M. persicum and M. kansasii type II, and between M. gastri and M. kansasii types I and IV.
Comparisons of the 277-bp ITS sequences from M. kansasii strains showed at most 85 and 98% similarities with the corresponding sequences from M. tuberculosis and other NTM species, respectively. Only sequences from M. kansasii type II and M. persicum were almost identical, sharing 99.2–100% similarity (Supplementary Table 2). The ITS sequence similarities within and between different M. kansasii subtypes fell under relatively wide ranges, i.e., 92.1–100% and 81.2–100%, respectively. The highest inter-subtype similarity values (>95%) were observed between three M. kansasii type I strains (K14, K19, and NLA00100521) and strains of M. kansasii type II. The type II-specific ITS sequences of those three type I strains accounted for the high intra-type heterogeneity (92.1–100% sequence similarity).
Sequence analysis of the 644-bp hsp65 gene fragments showed similarities of 90.3–92.3% between M. kansasii and M. tuberculosis, and 90.9–97.9% between M. kansasii and other NTM species, except M. persicum which shared 99.5–100% similarity with M. kansasii type II (Supplementary Table 3). Alignments of the hsp65 gene sequences from members of different M. kansasii subtypes yielded similarities of <98%. The only exception were three strains (K4, K14, and K19) of M. kansasii type I sharing up to 99.8% similarity with M. kansasii type II strains.
The results of the partial tuf (1,180 bp) gene analysis were similar to those obtained with the hsp65 gene (Supplementary Table 4). The tuf gene sequence similarities between M. kansasii and M. tuberculosis were 93.4% at most, while those between M. kansasii and other NTM species were always below 98%, except that sequences of M. kansasii type II and M. persicum were identical. The similarity indexes calculated for M. kansasii of different subtypes did not exceed 98.3%, except that two strains of type I (K14 and K19) had the same tuf sequences as M. kansasii type II strains.
Finally, alignments of the partial rpoB (3,439 bp) gene sequences found all M. kansasii strains to share <89% sequence similarity with M. tuberculosis and <96% similarity with NTM species, but again not M. persicum, whose sequences were identical or nearly identical with those of M. kansasii type II (Supplementary Table 5). The level of the rpoB gene sequence similarity between members of different M. kansasii subtypes was consistently below 98%, excluding two type I strains (K14 and K19), which displayed high similarity or identity with type II strains.
To better illustrate the phylogenetic relatedness of M. kansasii subtypes, phylogenetic trees inferred from five individual loci were constructed (Figures 3–7). A separate tree was created using the concatenated 16S rRNA, hsp65, and rpoB genes (Figure 8), since such an approach is known to increase considerably discrimination and robustness of the dendrogram analysis (Devulder et al., 2005). In all but one dendrograms, all M. kansasii strains could be spread into six highly supported (bootstrap values ≥ 93%) clusters, according to their subtype affiliation (Figures 4–8). The 16S rRNA gene-based dendrogram was different in that it contained no cluster specific for M. kansasii type IV. Two type IV strains clustered together with M. kansasii type I strains (Figure 3). Noteworthy, in the same cluster the type strain of M. gastri was placed. A feature, which was apparent across all the trees was that strains of M. kansasii type II clustered along with M. persicum. Moreover, there were four M. kansasii type I strains that branched within that cluster. Two of these strains (K14 and K19) were always present in the M. kansasii type II-M. persicum cluster, whereas another two belonged to that cluster only in the trees based on the ITS region (strain no. NLA00100521), 16S rRNA gene (NLA00100521), and hsp65 gene (strain no. K4). Having the type I-specific hsp65 gene sequence (sequevar I) and type II-specific ITS sequence (sequevar II), strain no. NLA00100521 represents the so-called intermediate type I (I/II), considered a transitional form between environmental type II and human-adapted type I (Iwamoto and Saito, 2005). Whereas, strains K14 and K19 can be identified as atypical type II (IIb) due to their type II ITS sequence and unique, yet most similar to type II, hsp65 sequences (Iwamoto and Saito, 2005). Captivatingly, strain K4 had the same unique hsp65 gene sequence, with all the other sequences being characteristic of type I. Thus, the strain represents the so far unreported variant of M. kansasii type I, which can be tentatively designated as atypical type I (Ib).

Figure 3. Phylogenetic tree based on 16S rRNA gene sequences, constructed using the Neighbor-Joining method. The bootstrap values were calculated from 1,000 replications. Bootstrap values are given at nodes. Strains of M. kansasii type IV (241/15, 1010001458) and M. gastri are shown boxed in solid and dashed lines, respectively. Strains of M. kansasii type I, as per WGS-based analysis, that clustered either within type I or II, upon single-gene or concatenated gene phylogenies are shown in bold. GenBank accession numbers for the sequences are parenthesized. Bar, 0.01 substitutions per nucleotide position.
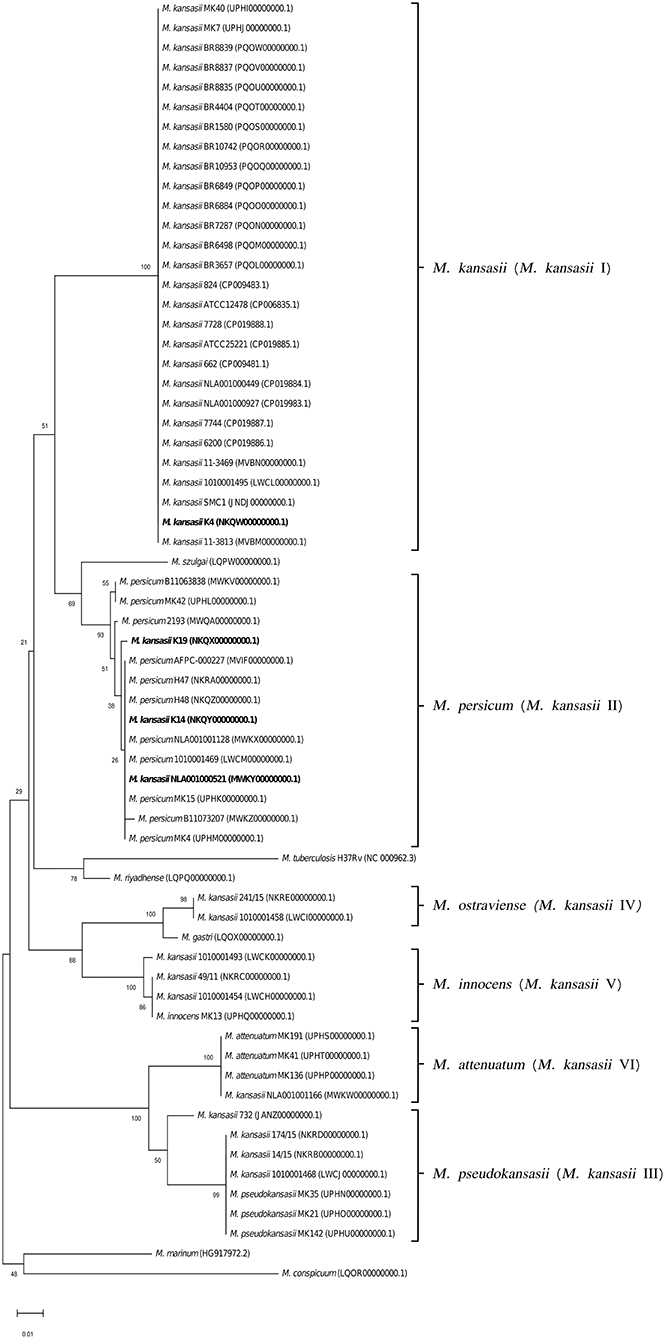
Figure 4. Phylogenetic tree based on ITS sequences constructed using the Neighbor-Joining method. The bootstrap values were calculated from 1,000 replications. Bootstrap values are given at nodes. Strains of M. kansasii type I, as per WGS-based analysis, that clustered either within type I or II, upon single-gene or concatenated gene phylogenies are shown in bold. GenBank accession numbers for the sequences are parenthesized. Bar, 0.01 substitutions per nucleotide position.
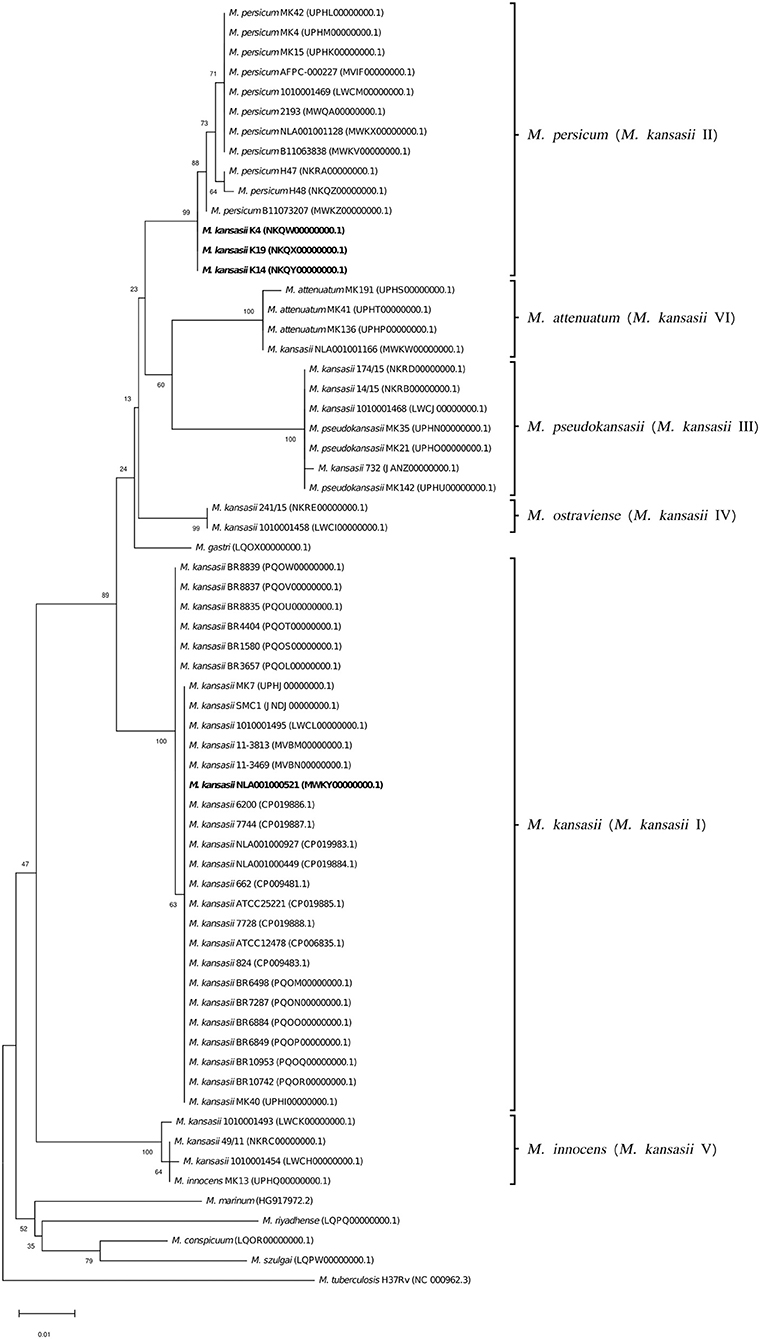
Figure 5. Phylogenetic tree based on hsp65 gene sequences constructed using the Neighbor-Joining method. The bootstrap values were calculated from 1,000 replications. Bootstrap values are given at nodes. Strains of M. kansasii type I, as per WGS-based analysis, that clustered either within type I or II, upon single-gene or concatenated gene phylogenies are shown in bold. GenBank accession numbers for the sequences are parenthesized. Bar, 0.01 substitutions per nucleotide position.
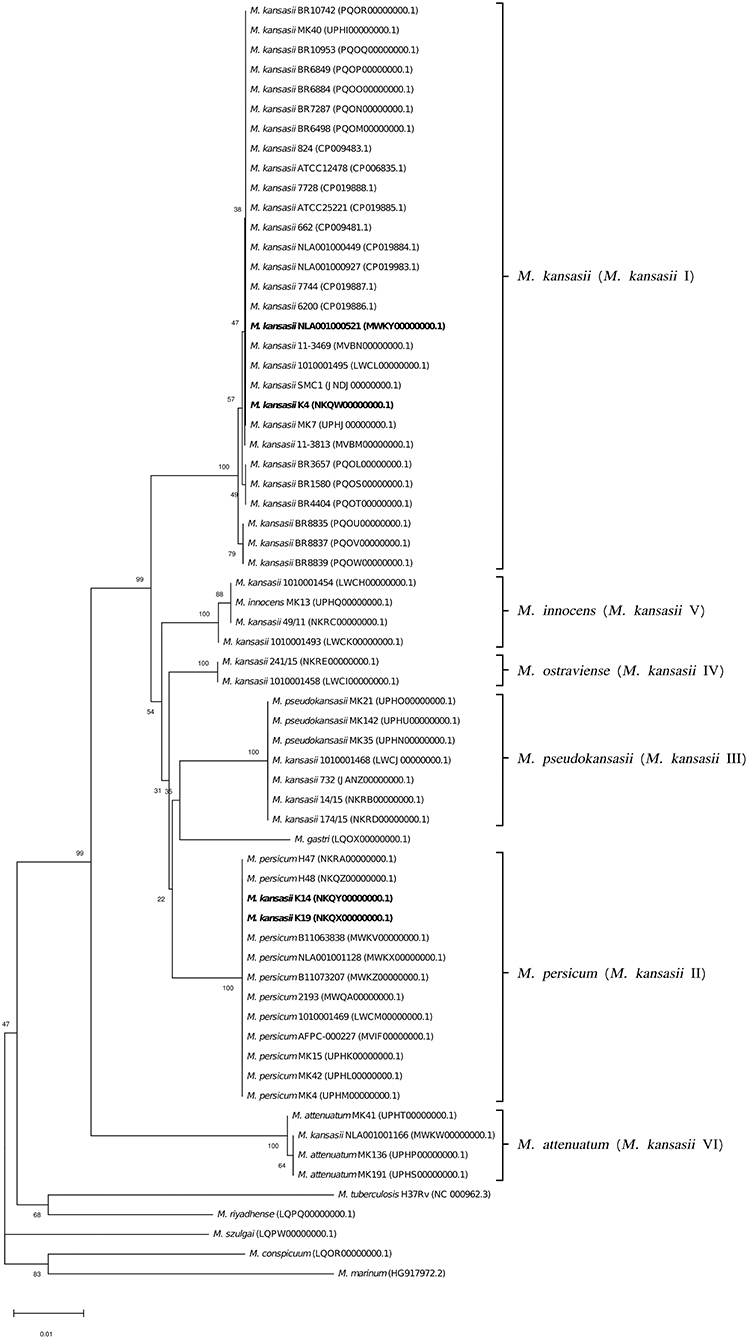
Figure 6. Phylogenetic tree based on tuf gene sequences constructed using the Neighbor-Joining method. The bootstrap values were calculated from 1,000 replications. Bootstrap values are given at nodes. Strains of M. kansasii type I, as per WGS-based analysis, that clustered either within type I or II, upon single-gene or concatenated gene phylogenies are shown in bold. GenBank accession numbers for the sequences are parenthesized. Bar, 0.01 substitutions per nucleotide position.
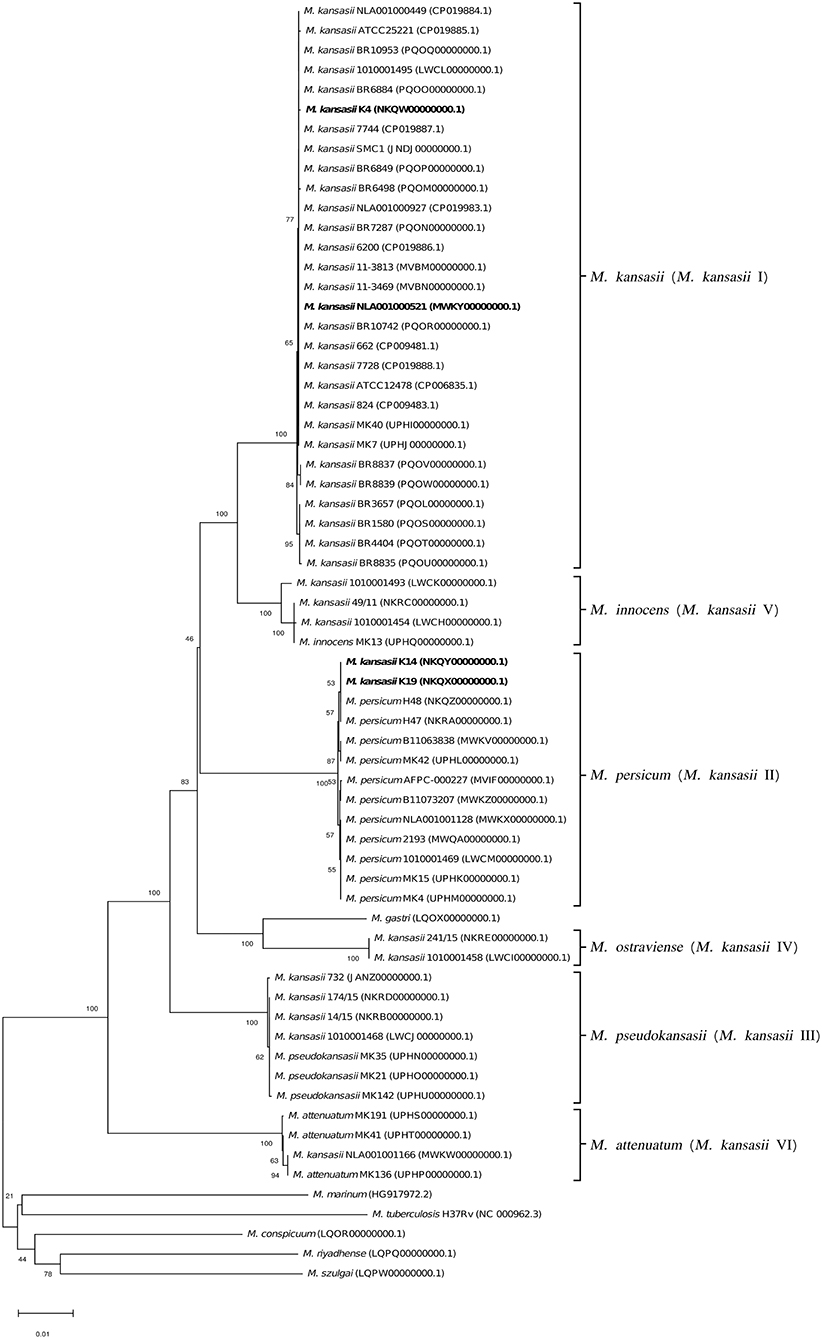
Figure 7. Phylogenetic tree based on rpoB sequences constructed using the Neighbor-Joining method. The bootstrap values were calculated from 1,000 replications. Bootstrap values are given at nodes. Strains of M. kansasii type I, as per WGS-based analysis, that clustered either within type I or II, upon single-gene or concatenated gene phylogenies are shown in bold. GenBank accession numbers for the sequences are parenthesized. Bar, 0.01 substitutions per nucleotide position.
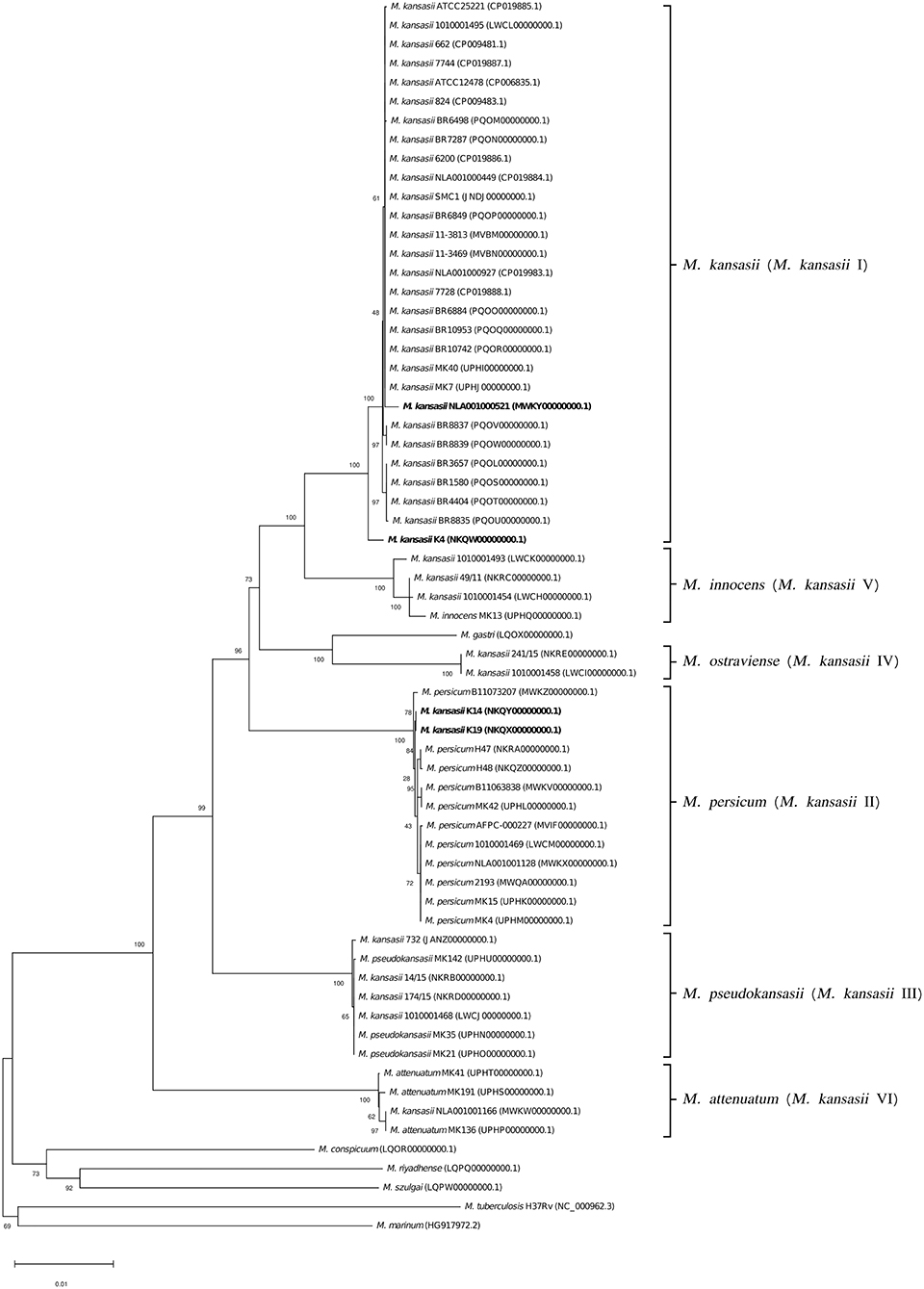
Figure 8. Phylogenetic tree based on concatenated 16S rRNA, hsp65, and rpoB gene sequences constructed using the Neighbor-Joining method. The bootstrap values were calculated from 1,000 replications. Bootstrap values are given at nodes. Strains of M. kansasii type I, as per WGS-based analysis, that clustered either within type I or II, upon single-gene or concatenated gene phylogenies are shown in bold. GenBank accession numbers for the sequences are parenthesized. Bar, 0.01 substitutions per nucleotide position.
Altogether, the results from the genome-wide comparisons demonstrated the six (I–VI) M. kansasii subtypes to represent distinct species. This was also the conclusion of the recent study by Tagini et al., who based their results upon ANI and GGD analysis of the genomes of 21 M. kansasii strains comprising all six subtypes (Tagini et al., 2019) (Twenty of these strains were used in the present work). Also, single- and multigene phylogenies, highly congruent between this and already published study, were indicative of species-level demarcations between M. kansasii subtypes. From these findings, Tagini et al. were first to propose new species designations, namely M. pseudokansasii, M. innocens, and M. attenuatum, replacing the former types III, V, and VI, respectively. The most prevalent type I was preserved under the ‘M. kansasii' designation. Whereas, M. kansasii type II was found, as in our study, conspecific with M. persicum. Therefore, we share the view of assigning this name to all M. kansasii type II strains. In fact, the conspecificity of M. persicum and M. kansasii type II would have been disclosed upon the original description of the species (M. persicum), if the authors had included M. kansasii type II in the genome-based comparative analysis (Shahraki et al., 2017).
The two M. kansasii type IV strains, analyzed in our study, fully satisfied the genomic criteria for a separate species. This was also implied by our predecessors, but in the absence of any type strain, they could not formally establish the species. Here, we propose a new species name, Mycobacterium ostraviense sp. nov., to accommodate M. kansasii type IV strains, with a strain no. 241/15, as a type. The description of this new species is given at the end of the article.
The taxonomic status of M. kansasii type VII remains an enigma. Neither the strain nor its genomic sequence is available, precluding any relevant phylogenetic analyses. This type was reported only once (Taillard et al., 2003), and given the similarity of its hsp65 RFLP banding patterns, which served as the only diagnostic means, to those of type III, it is plausible that type VII is a product of misidentification.
Since the mid-1990s, the PCR-RFLP analysis, based on single-copy, orthologous genes (hsp65, rpoB, and tuf ) has been widely used for the identification of a plethora of NTM species, including M. kansasii and its subtypes (Alcaide et al., 1997; Devallois et al., 1997; Kim et al., 2001; da Silva Rocha et al., 2002; Santin and Alcaide, 2003; Zhang et al., 2004; Kwenda et al., 2015; Bakuła et al., 2016). However, PCR-RFLP typing may not seldom produce misleading results. Single nucleotide polymorphisms can alter the recognition sites of the restriction enzymes and thus generate either patterns unidentifiable or corresponding to those of other species. Also, sequence analysis, even conducted on a combination of genes, may not resolve the species identity adequately. This is best illustrated in the already discussed strains of atypical type IIb, which despite sharing nearly 99% average nucleotide identity with type I, had their 16S rRNA, hsp65, and rpoB gene sequences, either individually or concatenated, more closely associated with type II. The type IIb strains would have been considered type II (M. persicum), if they had not been inspected at the whole genome-level. Thus, neither PCR-RFLP profiling nor single or multilocus sequencing allows unequivocal identification of M. persicum. A definite diagnosis should be supported by the genome-wide analysis. Still, the two type IIb strains, under this study, displayed the type I-specific hsp65 RFLP patterns, upon digestion with HaeIII (but not with BstEII, which was type II-specific). This feature can be exploited for differentiation between types I and II, if whole-genome sequencing is not affordable. Nevertheless, a new, robust genetic marker allowing for fast and accurate identification of all M. kansasii-derived species (former types I-VI), bypassing the need for whole-genome analysis, would be of great benefit. For this, a more in-depth, comparative analysis of the genomes of more strains representing the six M. kansasii-derived species, and other NTM species, will have to be undertaken.
There has been a continuing debate on how the differences between M. kansasii subtypes translate into pathogenicity. The prevailing view is that only types I and II are true human pathogens, with the latter having been associated with immunodeficiency, and HIV infection in particular, whereas all the remaining types are considered non-pathogenic, and their sporadic isolation from clinical samples has been interpreted as colonization or environmental contamination (Tortoli et al., 1994; Taillard et al., 2003). Indeed, M. kansasii type I is the most commonly detected among clinical isolates and the predominant cause of M. kansasii disease worldwide (Alcaide et al., 1997; Kim et al., 2001; Gaafar et al., 2003; Santin and Alcaide, 2003; Taillard et al., 2003; Zhang et al., 2004; da Silva Telles et al., 2005; Shitrit et al., 2006; Thomson et al., 2014; Kwenda et al., 2015; Bakuła et al., 2016). Infections attributable to M. kansasii type II are much rarer (Taillard et al., 2003; Zhang et al., 2004; Shitrit et al., 2006; Bakuła et al., 2016), and those caused by other types are almost unreported in the literature. Still, types III, IV, and VI have been recognized among clinical isolates (Alcaide et al., 1997; Picardeau et al., 1997; Santin and Alcaide, 2003; Thomson et al., 2014; Kwenda et al., 2015) with types IV and VI implicated in the disease (Santin and Alcaide, 2003; Thomson et al., 2014). Due to the paucity of isolations of M. kansasii other than type I, their clinical relevance remains obscure. Some new light on this problem may be shed by the findings of an international, multicenter investigation, currently in progress, on the global distribution of M. kansasii subtypes. So far, we have documented nine confirmed cases of M. kansasii disease, etiologically linked to either types III, IV, V or VI (Jagielski et al., data unpublished). In this context, the newly proposed species names for types V (M. innocens) and VI (M. attenuatum) may not reflect the true phenotype of those bacteria.
Searching for the genetic determinants of pathogenicity, a key genomic region associated with M. tuberculosis virulence, known as “region of difference 1” (RD1) was interrogated across the genomes of M. kansasii subtypes for its functional integrity (i.e., presence of RD1 genes). The RD1 encodes a secretory apparatus (ESX-1 type VII secretion system) responsible for exporting two highly potent antigens and virulence factors—the 6-kDa early secreted antigenic target (ESAT-6) and the 10-kDa culture filtrate protein (CFP-10) (Berthet et al., 1998). These proteins, encoded by the same operon, play a critical role in modulation of the host immune response through inhibition of phagosome maturation, cytosolic translocation of mycobacteria or granuloma formation (MacGurn and Cox, 2007; van der Wel et al., 2007; Volkman et al., 2010). The esat-6 (esxA) and cfp-10 (esxB) genes have also been demonstrated in some NTM species, including M. kansasii (types I-V), M. szulgai, M. marinum, and M. riyadhense (van Ingen et al., 2009). Furthermore, the ESAT-6/CFP-10-mediated translocation of bacilli into the cytosol has been proven to occur in M. kansasii type I, but not in M. kansasii type V (Houben et al., 2012).
Our analysis showed the presence of six RD1 genes (rv3871-5 and rv3877), including the ESAT-6 and CFP-10-coding genes and two other genes (rv3871 and rv3877) coding for the essential components of the ESX-1 secretion system, in all types of M. kansasii and other NTM species (Table 5). Only single strains of M. kansasii type I, M. marinum, and M. szulgai were devoid of the rv3872 gene coding for PE35, conjectured to play a role in the regulation of esxB/A expression (Brodin et al., 2006).
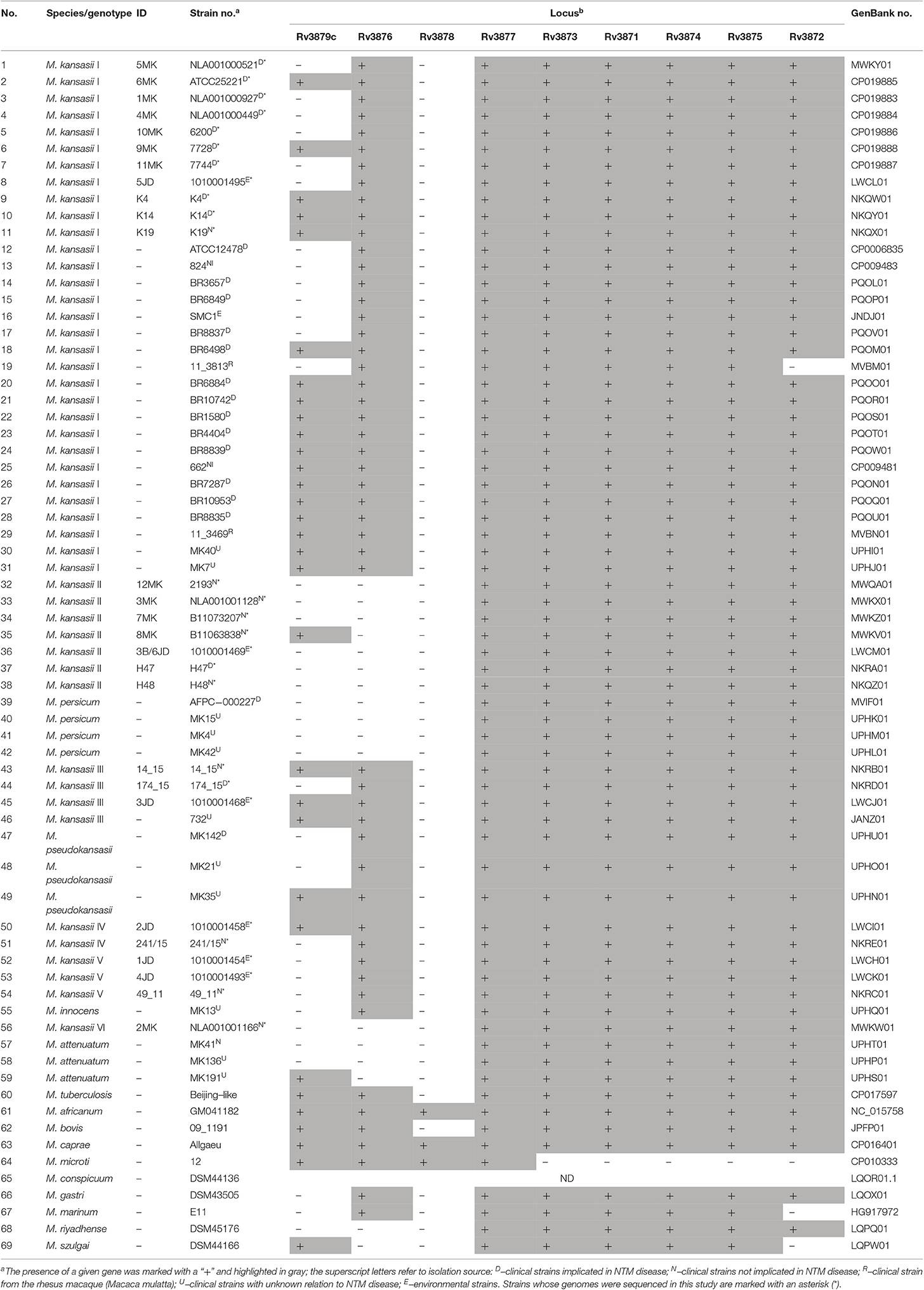
Table 5. Distribution of region of difference 1 (RD1) genes among M. kansasii strains and other mycobacterial outgroup species.
The rv3876 gene was demonstrated in all M. kansasii types except for types II (M. persicum) and VI (M. attenuatum). Neither it was present in M. szulgai and M. riyadhense. The protein encoded by this gene is an ESX-1 secretion-associated protein EspI. It was shown that inactivation of the rv3876 gene did not impair secretion of ESAT-6 (Brodin et al., 2006). More recently, however, EspI was found to negatively regulate the ESX-1 secretion system in M. tuberculosis, in response to low cellular ATP levels. EspI was thus hypothesized to play a possible role during the latent phase of infection (Zhang et al., 2014).
The rv3879c gene, coding for another ESX-1 secretion-associated protein EspK, was variably distributed among M. kansasii strains. It was detected in half of the strains of types I and III, while being absent in all but one strains of types II and VI, and all type V strains. EspK seems not to be involved in virulence, since the rv3879c homolog deletion mutant of M. bovis was not attenuated in the guinea pig model (Inwald et al., 2003). Moreover, similar to rv3876, inactivation of the EspK-coding gene did not abolish ESAT-6 secretion (Brodin et al., 2006). Contrastingly, EspK of M. marinum was found crucial for the ESX-1-mediated secretion of EspB (Rv3881c), required for virulence and growth in macrophages (McLaughlin et al., 2007). It is thus conceivable that EspK may influence the pathogenicity also in M. kansasii.
Collectively, based on the distribution of RD1 genes, neither of the M. kansasii types could be categorized as being more or less pathogenic, given that all genes essential for the functioning of the ESX-1 secretion machinery were uniformly present in M. kansasii strains, and that the absence of certain genes was reported in both clinically relevant and neutral strains. To explore more in depth the genetic background of virulence in M. kansasii, more advanced, functional studies should be performed, with a focus not only on the RD1 genes but several other genes flanking that cluster, which together form the “extended RD1” region. Furthermore, we cannot exclude that the pathogenic and non-pathogenic M. kansasii strains differ in terms of expression of the RD1 genes or activity of their proteins. More RD1-targeted experimental investigations are required to validate such scenarios.
Analysis of other than RD1 regions of deletions (RD2-14) did not show any consistent (i.e., shared across all strains of a given species) species-specific pattern of RD genes (Supplementary Data Sheet 2). It was noteworthy, however, that the only two genes found in the RD3 locus were present in either of the two most pathogenic M. kansasii types I and II. The rv1577 gene occurred in more than 90% of M. kansasii type I and almost 73% of M. kansasii type II. Whereas, the rv1586 gene was demonstrated exclusively in M. kansasii type I, at a frequency of nearly 81%. Likewise, only M. kansasii types I and II harbored genes of the RD11 locus. The rv2651 gene was present in 90 and 73% of M. kansasii type I and II, respectively. The rv2646 was evidenced in slightly more than 60% of M. kansasii type I. Interestingly, both RD3 and RD11 represent phage inserts within the M. tuberculosis genome, and are thought to generate antigenic variation (Ahmed et al., 2007).
It has been canonically accepted, upon description of new species, to provide a detailed phenotypic characterization. However, in the era of genomic-based bacterial taxonomy, the significance of the phenotype has much eroded and the use of conventional biochemical testing has been increasingly abandoned. The algorithm for routine differential diagnostics of NTM species should obligatorily include only growth rate and pigment production (Tortoli et al., 2017).
For closely-related species, the diagnostic value of phenotyping is virtually negligible, as demonstrated also in this study (Table 6). All strains, irrespective of subtype (species), were almost invariably photochromogenic, niacin-negative, and grew at 25 and 37°C, but not at 45°C, in the presence of 5% (w/v) NaCl or on MacConkey agar without crystal violet. They were all resistant to thiophene-2-carboxylic acid hydrazide (TCH) and presented a strong catalase activity, but none exhibited pyrazynamidase activity or arylsulfatase at 3 days. Some variability was observed when testing for nitrate and tellurite reduction, Tween 80 hydrolysis, and urease activity. The only marked difference between the subtypes (species) was that strains of M. ostraviense (formerly M. kansasii type IV) were, unlike all other species, unable to reduce nitrate and that their catalase was heat-liable. Whether these features are stable within the species need to be verified on a larger set of strains. Interestingly, both these features are typical for M. gastri (Kent and Kubica, 1985), with which M. ostraviense shares the highest genetic similarity, as evidenced upon whole-genome analysis. Mycobacterium gastri, a casual resident of human stomach and only exceptionally pathogenic (Velayati et al., 2005), is easily distinguishable from all M. kansasii-derived species, as it is non-photochromogenic.
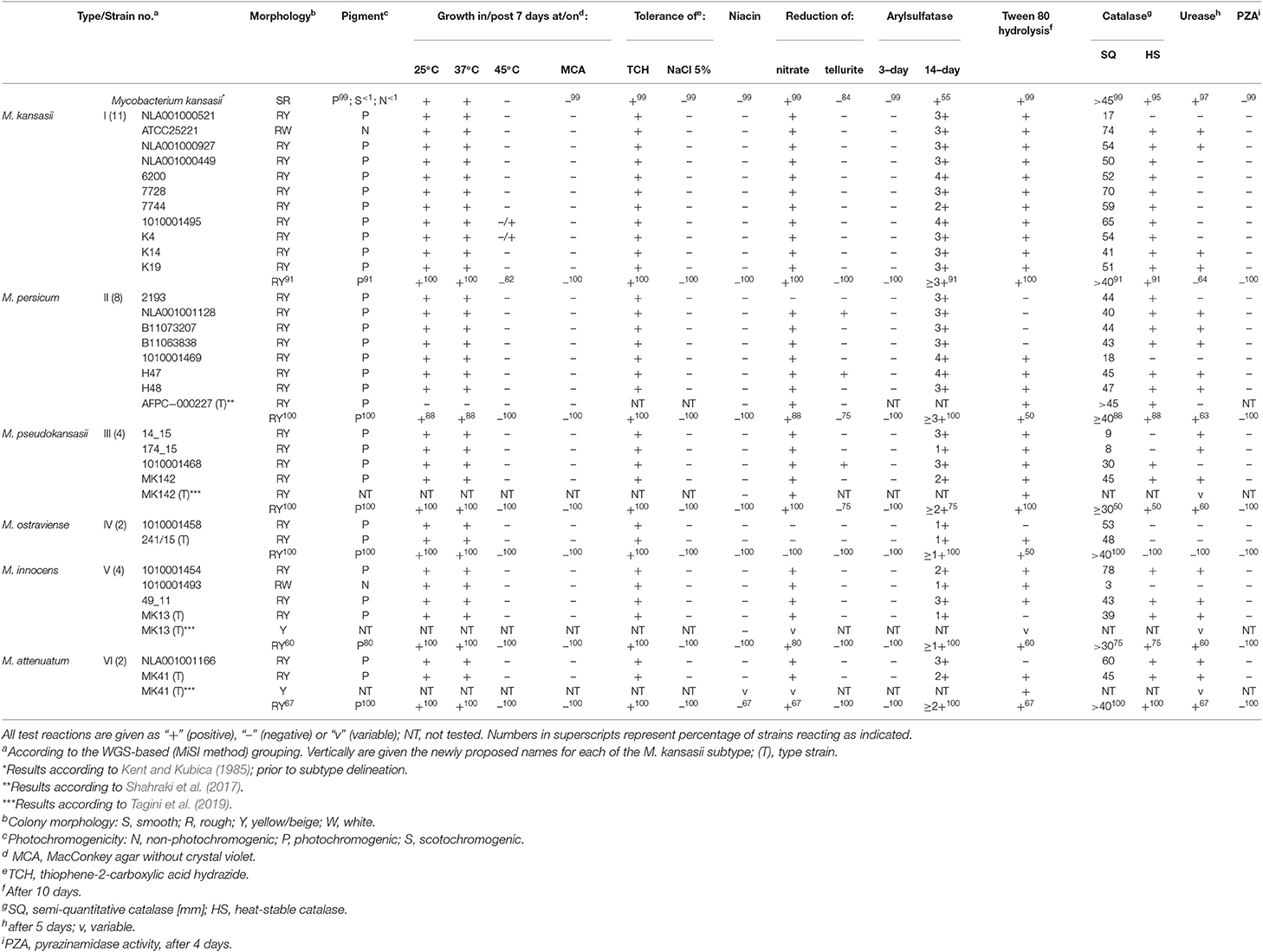
Table 6. Phenotypic characteristics of M. kansasii strains under the study.
Finally, drug susceptibility profiles of 30 mycobacterial strains, including type strains of three newly established species, by Tagini et al. (2019), were compared within and between the species (former M. kansasii types) (Table 7). Shortly, all strains were susceptible to RIF, RFB, AMK, SXT, MFX, LZD, and CLR (except one CLR-resistant strain of M. kansasii). Of these drugs, only RIF and CLR showed interspecies differences in their activity, with the MICs for M. kansasii and M. persicum slightly higher than for other M. kansasii-derived species. More than 80% of strains were resistant to EMB. Among these, were all strains of M. kansasii (former type I), M. persicum (II), and M. ostraviense (IV). Single strains of M. kansasii, M. pseudokansasii, and M. attenuatum were resistant to CIP. The MICs of STR and DOX varied widely (<0.5–16 mg/L vs. 1–>16 mg/L), yet the highest values (16 vs. >16 mg/L) were observed only for strains of M. kansasii, M. persicum, and M. attenuatum. The INH and ETO MICs were low, and within relatively narrow ranges (<0.25–2 mg/L vs. <0.3–0.6 mg/L). These findings confirm the key observations from previous studies, on the susceptibility of M. kansasii strains to RIF and their high resistance to EMB (da Silva Telles et al., 2005; Wu et al., 2009; Shahraki et al., 2017; Bakuła et al., 2018c). This, confronted with the ATS recommendation of a three-drug (INH-RIF-EMB) regimen for the treatment of M. kansasii disease, speaks for exclusion of EMB and its replacement with other potent drug, such as moxifloxacin.
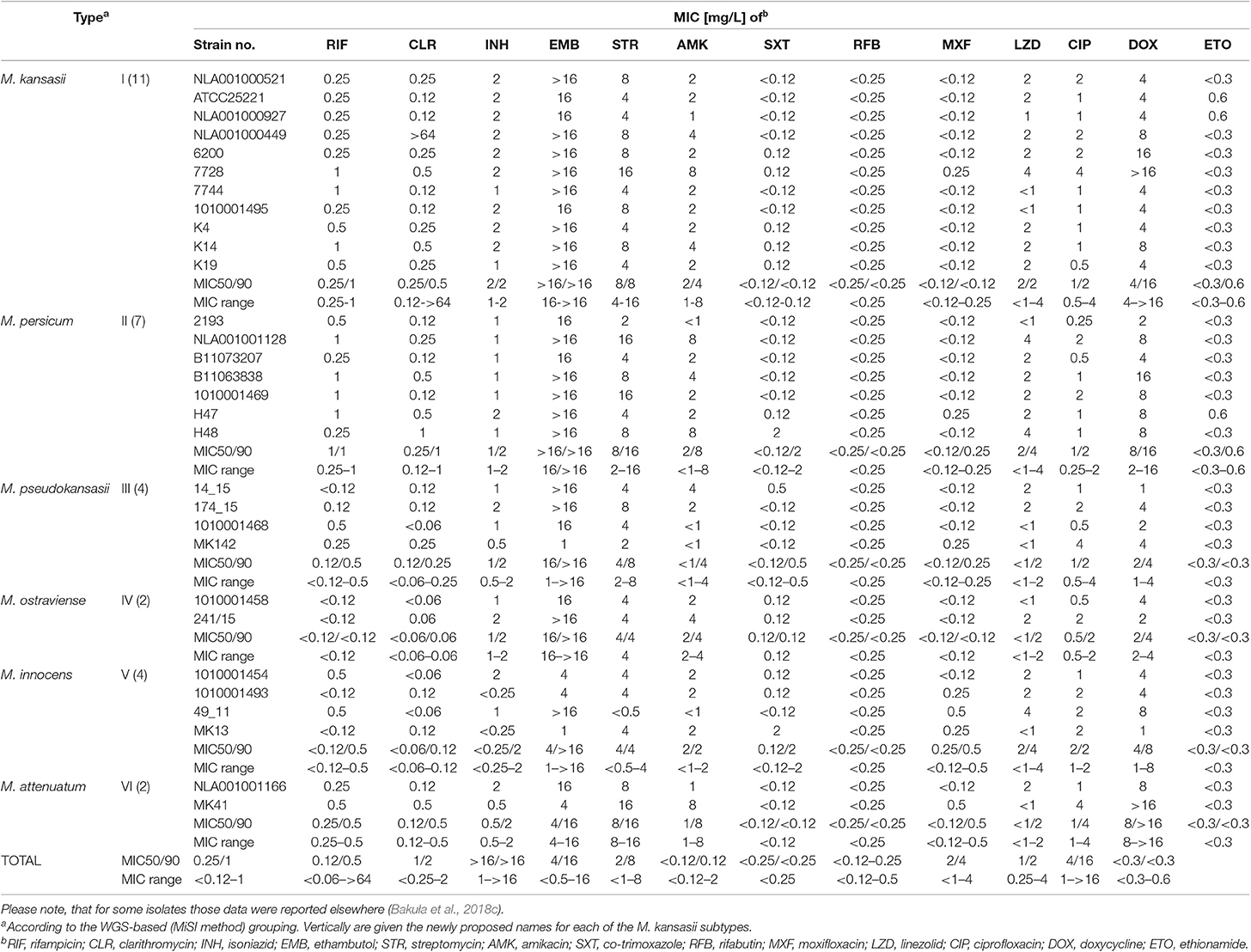
Table 7. Drug susceptibility profiles of M. kansasii strains under the study.
In conclusion, the present paper updates and extends the findings of earlier investigation on the taxonomy of M. kansasii. Not only does it further substantiate the delineation of new species from the M. kansasii group to replace the former subtypes I–VI, but consolidates the position of five of the so erected species, and provides a description of the sixth one, M. ostraviense, a successor of the subtype IV. By showing a close genetic relatedness, a monophyletic origin, and overlapping phenotypes, our findings support the recognition of the M. kansasii complex (MKC), accommodating all M. kansasii-derived species and M. gastri. Neither of the most commonly used taxonomic markers can accurately distinguish all the MKC species. Likewise, no species-specific phenotypic characteristics exist that would allow identification of the species, except the non-photochromogenicity of M. gastri. In the context of the previously proposed polyphasic strategy in resolving species boundaries and their interrelatedness, FAME (fatty acid methyl ester) analysis, as an adjunct typing method, might be useful (Saini et al., 2009). However, preparatory techniques for FAME analysis are typically time-consuming, laborious, and material-intensive. Furthermore, chromatography-dedicated facilities require investment in instrumentation and training, and despite their services being offered by universities and other centres, they are often less accessible than sequencing facilities, even in the developing countries.
To distinguish, most reliably, between the MKC species, and between M. kansasii and M. persicum in particular, whole-genome-based approaches should be applied.
Since no clear differences in the repertoire of the virulence-associated RD1 genes have been observed among the M. kansasii-derived species, the pathogenic capacity of each of these species can only be speculated based on their prevalence among the clinically relevant population. Large-scale molecular epidemiological studies are needed to gain a better understanding of the clinical significance and pathobiology of the MKC species.
Description of Mycobacterium ostraviense sp. nov. Jagielski and Ulmann
Mycobacterium ostraviense [os.tra.vi.en'se. N.L. neut. adj. ostraviense pertaining to Ostravia, the Latin name of Ostrava, a city in the north-east of the Czech Republic where one of the strain was isolated].
The species name refers to the former M. kansasii subtype IV. Mature colonies, of rough surface and photochromogenic, are observed on Löwenstein-Jensen medium after more than 7 days of incubation at 37°C (Supplementary Figure 1). No growth occurs at 45°C and on the media containing TCH or 5% (w/v) NaCl. Similar to other MKC species, it tests positive for the semi-quantitative catalase (>45 mm) and 14-day arylsulfatase activity, while negative for niacin accumulation, tellurite reduction, and urease and pyrazynamidase activities. Unlike to other M. kansasii-derived species but similar to M. gastri, it does not reduce nitrates, nor it produces a heat-stable (68°C) catalase. Strongly resistant to ethambutol (> 16 mg/L) but susceptible to amikacin, clarithromycin, co-trimoxazole, linezolid, fluoroquinolones, and rifamycins.
The species has the same 16S rRNA sequences as M. kansasii (former subtype I) and M. gastri, yet it displays unique sequences at the hsp65, tuf, and rpoB genes, and the ITS locus. At the genomic level, it is most closely related to M. gastri, with pairwise ANI and GGD values of 95.2 and 0.056, respectively.
The type strain, 241/15T was isolated from a sputum of a patient with no NTM disease, based in Karviná, near Ostrava, in the Moravian-Silesian Region of the Czech Republic. The type strain has been deposited in the Leibniz Institute German Collection of Microorganisms and Cell Cultures (DSMZ; Braunschweig, Germany) under the accession number DSM 110538.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Ethics Statement
Analyses were based on data that did not contain any sensitive personal information. Therefore, informed consent and ethical approval were not required in like with local legislation.
Author Contributions
TJ conceptualized and supervised the study, provided the funding, organized and integrated the data, and wrote the manuscript. PB, JL, and DS performed bioinformatic analyses including AF-ANI, GGD, and phylogenetic tree analysis. ZB performed culturing, subtyping, and phenotypic profiling of M. kansasii strains. BM carried out analysis on regions of difference 1-14 (RD1-14) with a homemade script Diffind. AB carried out DNA isolations for whole-genome sequencing. JD analyzed the results on the distribution of the RD1-14 genes in M. kansasii genomes. MD constructed the core-genome phylogenies. LP performed drug susceptibility testing. JI provided 13 M. kansasii strains of subtypes I-VI and critically reviewed the manuscript. MZ-D co-performed phenotypic assays.
Funding
This work was supported by the National Centre for Research and Development grant (to: TJ) through the LIDER Programme under contract no. LIDER/044/457/L-4/12/NCBR/2013. DS, JL, and BM were supported by the Polish Ministry of Science and Higher Education grant under contract no. DIR/WK/2017/2018/01-1.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are indebted to Aleksandra Safianowska (Department of Internal Medicine, Pulmonology, and Allergology, Warsaw Medical University, Warsaw, Poland), Won-Jung Koh (Division of Pulmonary and Critical Care Medicine; Department of Medicine, Samsung Medical Center, Sungkyunkwan University School of Medicine, South Korea), Vit Ulmann (Institute of Public Health, Ostrava, Czech Republic), Miren J. Unzaga Barañano (Clinical Microbiology and Infection Control, Basurto Hospital, Bilbao, Spain), Joanna Humiecka (Hospital of Infectious Diseases in Warsaw, Poland), and Katharina Kranzer (National Reference Laboratory for Mycobacteria, Research Center Borstel, Borstel, Germany) for their kind providing some of M. kansasii strains used in this study. Special appreciation is extended to Fernando Alcaide and David Rodriguez-Temporal (Department of Pathology and Experimental Therapy, Universitat de Barcelona, Hospitalet de Llobregat, Spain) for their kind reviewing of the manuscript and valuable comments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02918/full#supplementary-material
Supplementary Tables 1–6 are published inside Supplementary Data Sheet 1.
Supplementary Table 1. Pairwise similarities (%) between 16S rRNA sequences.
Supplementary Table 2. Pairwise similarities (%) between ITS sequences.
Supplementary Table 3. Pairwise similarities (%) between hsp65 sequences.
Supplementary Table 4. Pairwise similarities (%) between tuf sequences.
Supplementary Table 5. Pairwise similarities (%) between rpoB sequences.
Supplementary Table 6. Strains (genomes) used in different genetic analyses.
Supplementary Figure 1. Macro- and micromorphology of colonies of four recently established species within the M. kansasii complex. Colonies photographed after 2 weeks of growth at 37°C on Löwenstein-Jensen medium, and additional 72 h post-light exposure (upper row of images). Cell morphological patterns upon Ziehl-Neelsen stain (magnification, x100), and scanning electron microscopy (magnification, x6,000). Scale bar 2 μM is applicable for M. pseudokansasii, M. ostraviense, and M. innocens, while scale bar 3 μM applies to M. attenuatum.
Supplementary Data Sheet 2. Distribution of region of difference 2-14 (RD2-14) genes among M. kansasii strains and other mycobacterial outgroup species.
References
Abed, Y., Bollet, C., and de Micco, P. (1995). Demonstration of Mycobacterium kansasii species heterogeneity by the amplification of the 16S-23S spacer region. J. Med. Microbiol. 42, 112–114. doi: 10.1099/00222615-43-2-156
Ahmed, N., Saini, V., Raghuvanshi, S., Khurana, J. P., Tyagi, A. K., Tyagi, A. K., et al. (2007). Molecular analysis of a leprosy immunotherapeutic bacillus provides insights into Mycobacterium evolution. PLoS ONE 2:e968. doi: 10.1371/journal.pone.0000968
Alcaide, F., Richter, I., Bernasconi, C., Springer, B., Hagenau, C., Schulze-Röbbecke, R., et al. (1997). Heterogeneity and clonality among isolates of Mycobacterium kansasii: implications for epidemiological and pathological studies. J. Clin. Microbiol. 35, 1959–1964.
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Auch, A. F., Klenk, H. P., and Göker, M. (2010). Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand. Genomic Sci. 2, 142–148. doi: 10.4056/sigs.541628
Bakuła, Z., Brzostek, A., Borówka, P., Zaczek, A., Szulc-Kiełbik I, Podpora, A., Parniewski, P., et al. (2018a). Molecular typing of Mycobacterium kansasii using pulsed-field gel electrophoresis and a newly designed variable-number tandem repeat analysis. Sci. Rep. 8:e4462. doi: 10.1038/s41598-018-21562-z
Bakuła, Z., Kościuch, J., Safianowska, A., Proboszcz, M., Bielecki, J., van Ingen, J., et al. (2018b). Clinical, radiological and molecular features of Mycobacterium kansasii pulmonary disease. Respir. Med. 139, 91–100. doi: 10.1016/j.rmed.2018.05.007
Bakuła, Z., Modrzejewska, M., Pennings, L., Proboszcz, M., Safianowska, A., Bielecki, J., et al. (2018c). Drug susceptibility profiling and genetic determinants of drug resistance in Mycobacterium kansasii. Antimicrob Agents Chemother. C 62, e01788–e01717. doi: 10.1128/AAC.01788-17
Bakuła, Z., Modrzejewska, M., Safianowska, A., van Ingen, J., Proboszcz, M., Bielecki, J., et al. (2016). Proposal of a new method for subtyping of Mycobacterium kansasii based upon PCR restriction enzyme analysis of the tuf gene. Diagn. Microbiol. Infect. Dis. 84, 318–321. doi: 10.1016/j.diagmicrobio.2015.12.009
Berthet, F. X., Rasmussen, P. B., Rosenkrands, I., Andersen, P., and Gicquel, B. (1998). A Mycobacterium tuberculosis operon encoding ESAT-6 and a novel low-molecular-mass culture filtrate protein (CFP-10). Microbiology 144, 3195–3203. doi: 10.1099/00221287-144-11-3195
Brodin, P., Majlessi, L., Marsollier, L., de Jonge, M. I., Bottai, D., Demangel, C., et al. (2006). Dissection of ESAT-6 system 1 of Mycobacterium tuberculosis and impact on immunogenicity and virulence. Infect. Immun. 74, 88–98. doi: 10.1128/IAI.74.1.88-98.2006
Brosch, R., Gordon, S. V., Marmiesse, M., Brodin, P., Buchrieser, C., Eiglmeier, K., et al. (2002). A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc. Natl. Acad. Sci. U.S.A. 99, 3684–3689. doi: 10.1073/pnas.052548299
Buhler, V. B., and Pollak, A. (1953). Human infection with atypical acid-fast organisms; report of two cases with pathologic findings. Am. J. Clin. Pathol. 23, 363–374. doi: 10.1093/ajcp/23.4.363
Butler, W. R., Floyd, M. M., Silcox, V., Cage, G., Desmond, E., Duffey, P. S., et al. (1996). Standardized Method for HPLC Identification of Mycobacteria. HPLC users group in cooperation with Centers for Disease Control and Prevention. Atlanta, GA: U.S. Public Health Service, CDC.
Chen, Y. P., Yen, Y. S., Chen, T. Y., Yen, C. L., Shieh, C. C., and Chang, K. C. (2008). Systemic Mycobacterium kansasii infection mimicking peripheral T-cell lymphoma. APMIS 116, 850–858. doi: 10.1111/j.1600-0463.2008.00935.x
CLSI; Clinical and Laboratory Standards Institute (2011). Susceptibility Testing of Mycobacteria, Nocardiae, and Other Aerobic Actinomycetes; Approved Standard, 2nd Edn. CLSI document M24-A2. Clinical and Laboratory Standards Institute, Wayne, IL.
da Silva Rocha, A., Werneck Barreto, A. M., Dias Campos, C. E., Villas-Bôas da Silva, M., Fonseca, L., Saad, M. H., et al. (2002). Novel allelic variants of Mycobacteria isolated in Brazil as determined by PCR-restriction enzyme analysis of hsp65. J. Clin. Microbiol. 40, 4191–4196. doi: 10.1128/JCM.40.11.4191-4196.2002
da Silva Telles, M. A., Chimara, E., Ferrazoli, L., and Riley, L. W. (2005). Mycobacterium kansasii: antibiotic susceptibility and PCR-restriction analysis of clinical isolates. J. Med. Microbiol. 54, 975–979. doi: 10.1099/jmm.0.45965-0
Devallois, A., Goh, K. S., and Rastogi, N. (1997). Rapid identification of mycobacteria to species level by PCR-restriction fragment length polymorphism analysis of the hsp65 gene and proposition of an algorithm to differentiate 34 mycobacterial species. J. Clin. Microbiol. 35, 2969–2973.
Devulder, G., Pérouse de Montclos, M., and Flandrois, J. P. (2005). A multigene approach to phylogenetic analysis using the genus Mycobacterium as a model. Int. J. Syst. Evol. Microbiol. 55, 293–302. doi: 10.1099/ijs.0.63222-0
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
Falkinham, J. O. (1996). Epidemiology of Infection by nontuberculous mycobacteria. Clin. Microbiol. Rev. 9, 177–215. doi: 10.1128/CMR.9.2.177
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x
Gaafar, A., Unzaga, M. J., Cisterna, R., Clavo, F. E., Urra, E., Ayarza, R., et al. (2003). Evaluation of a modified single-enzyme amplified-fragment length polymorphism technique for fingerprinting and differentiating of Mycobacterium kansasii type I isolates. J. Clin. Microbiol. 41, 3846–3850. doi: 10.1128/JCM.41.8.3846-3850.2003
Griffith, D. E., Aksamit, T., Brown-Elliott, B. A., Catanzaro, A., Daley, C., Gordin, F., et al. (2007). ATS Mycobacterial Diseases Subcommittee; American Thoracic Society; Infectious Disease Society of America. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am. J. Respir. Crit. Care Med. 175, 367–416. doi: 10.1164/rccm.200604-571ST
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98.
Hermans, P. W., van Soolingen, D., and van Embden, J. D. (1992). Characterization of a major polymorphic tandem repeat in Mycobacterium tuberculosis and its potential use in the epidemiology of Mycobacterium kansasii and Mycobacterium gordonae. J. Bacteriol. 174, 4157–4165. doi: 10.1128/jb.174.12.4157-4165.1992
Hoefsloot, W., van Ingen, J., Andrejak, C., Angeby, K., Bauriaud, R., Bemer, P., et al. (2013). The geographic diversity of nontuberculous mycobacteria isolated from pulmonary samples: a NTM-NET collaborative study. Eur. Respir. J. 42, 1604–1613. doi: 10.1183/09031936.00149212
Horsburgh, C. R. Jr., and Selik, R. M. (1989). The epidemiology of disseminated nontuberculous mycobacterial infection in the acquired immunodeficiency syndrome (AIDS). Am. Rev. Respir. Dis. 139, 4–7. doi: 10.1164/ajrccm/139.1.4
Houben, D., Demangel, C., van Ingen, J., Perez, J., Baldeón, L., Abdallah, A. M., et al. (2012). ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell. Microbiol. 14, 1287–1298. doi: 10.1111/j.1462-5822.2012.01799.x
Iinuma, Y., Ichiyama, S., Hasegawa, Y., Shimokata, K., Kawahara, S., and Matsushima, T. (1997). Large-restriction-fragment analysis of Mycobacterium kansasii genomic DNA and its application in molecular typing. J. Clin. Microbiol. 35, 596–599.
Inwald, J., Jahans, K., Hewinson, R. G., and Gordon, S. V. (2003). Inactivation of the Mycobacterium bovis homologue of the polymorphic RD1 gene Rv3879c (Mb3909c) does not affect virulence. Tuberculosis 83, 387–393. doi: 10.1016/j.tube.2003.08.018
Iwamoto, T., and Saito, H. (2005). Comparative study of two typing methods hsp65 PRA and ITS sequencing revealed a possible evolutionary link between Mycobacterium kansasii type I and II isolates. FEMS Microbiol. Lett. 254, 129–133. doi: 10.1111/j.1574-6968.2005.00013.x
Jukes, T. H., and Cantor, C. R. (1969). “Evolution of protein molecules,” in Mammalian Protein Metabolism, ed H. N. Munro (New York, NY: Academic Press), 21–132.
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kent, P. T., and Kubica, G. P. (1985). Public Health Mycobacteriology. A Guide for the Level III Laboratory. Atlanta, GA: Centers for Disease Control.
Kim, B. J., Lee, K. H., Park, B. N., Kim, S. J., Bal, G. H., Kim, S. J., et al. (2001). Differentiation of mycobacterial species by PCR-restriction analysis of DNA (343 base pairs) of the RNA polymerase gene (rpoB). J. Clin. Microbiol. 39, 2102–2109. doi: 10.1128/JCM.39.6.2102-2109.2001
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kwenda, G., Churchyard, G. J., Thorrold, C., Heron, I., Stevenson, S., Duse, A. G., et al. (2015). Molecular characterization of clinical and environmental isolates of Mycobacterium kansasii isolates from South African gold mines. J. Water Health 13, 190–202. doi: 10.2166/wh.2014.161
Liao, C. H., Lai, C. C., Ding, L. W., Hou, S. M., Chiu, H. C., Chang, S. C., et al. (2007). Skin and soft tissue infection caused by non-tuberculous mycobacteria. Int. J. Tuberc. Lung Dis. 11, 96–102.
LPSN database (2019). List of Prokaryotic Names With Standing in Nomenclature. Available online at: http://www.bacterio.net/ (accessed August 24, 2019).
MacGurn, J. A., and Cox, J. S. (2007). A genetic screen for Mycobacterium tuberculosis mutants defective for phagosome maturation arrest identifies components of the ESX-1 secretion system. Infect. Immun. 75, 2668–2678. doi: 10.1128/IAI.01872-06
Manten, A. (1957). Antimicrobial susceptibility and some other properties of photochromogenic mycobacteria associated with pulmonary disease. Antonie van Leeuwenhoek. J. Microbiol. Serol. 23, 357–363. doi: 10.1007/BF02545888
Marciniak, B., Borówka, P., and Strapagiel, D. (2017). DIFFIND – The Sequence Difference Finder. Available online at: https://github.com/BiobankLab/DIFFIND
Marras, T. K., and Daley, C. L. (2004). A systematic review of the clinical significance of pulmonary Mycobacterium kansasii isolates in HIV infection. J. Acquir. Immune Defic. Syndr. 36, 883–889. doi: 10.1097/00126334-200408010-00001
Matveychuk, A., Fuks, L., Priess, R., Hahim, I., and Shitrit, D. (2012). Clinical and radiological features of Mycobacterium kansasii and other NTM infections. Respir. Med. 106, 1472–1477. doi: 10.1016/j.rmed.2012.06.023
McLaughlin, B., Chon, J. S., MacGurn, J. A., Carlsson, F., Cheng, T. L., Cox, J. S., et al. (2007). A mycobacterium ESX-1-secreted virulence factor with unique requirements for export. PLoS Pathog. 3:e105. doi: 10.1371/journal.ppat.0030105
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., and Göker, M. (2013a). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60
Meier-Kolthoff, J. P., Göker, M., Spröer, C., and Klenk, H. P. (2013b). When should a DDH experiment be mandatory in microbial taxonomy? Arch. Microbiol. 195, 413–418. doi: 10.1007/s00203-013-0888-4
Middlebrook, G. (1956). Diagnostic and biological problems of isoniazid-resistant tubercle bacilli. Bull. Un. Int. Tuberc. 26, 179–205.
Moon, S. M., Park, H. Y., Jeon, K., Kim, S. Y., Chung, M. J., Huh, H. J., et al. (2015). Clinical significance of Mycobacterium kansasii isolates from respiratory specimens. PLoS ONE 10:e0139621. doi: 10.1371/journal.pone.0139621
Murugaiyan, J., Lewin, A., Kamal, E., Bakuła, Z., van Ingen, J., Ulmann, V., et al. (2018). MALDI spectra database for rapid discrimination and subtyping of Mycobacterium kansasii. Front. Microbiol. 9:e587. doi: 10.3389/fmicb.2018.00587
Nurk, S., Bankevich, A., Antipov, D., Gurevich, A. A., Korobeynikov, A., Lapidus, A., et al. (2013). “Assembling genomes and mini-metagenomes from highly chimeric reads,” in Annual International Conference on Research in Computational Molecular Biology (Berlin; Heidelberg Springer).
Park, S. Y., Lee, G. R., Min, J. W., Jung, J. Y., Jeon, Y. D., Shin, H. S., et al. (2012). A case of Mycobacterium kansasii lymphadenitis in HIV-infected patient. Infect. Chemother. 44, 526–529. doi: 10.3947/ic.2012.44.6.526
Picardeau, M., Prod'hom, G., Raskine, L., LePennec, M. P., and Vincent, V. (1997). Genotypic characterization of five subspecies of Mycobacterium kansasii. J. Clin. Microbiol. 35, 25–32.
Plikaytis, B. B., Plikaytis, B. D., Mitchell, A., Yakrus, W., Butler, R., Woodley, C. L., et al. (1992). Differentiation of slowly growing Mycobacterium species, including Mycobacterium tuberculosis, by gene amplification and restriction fragment length polymorphism analysis. J. Clin. Microbiol. 30, 1815–1822.
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS ONE. 5:e9490. doi: 10.1371/journal.pone.0009490
Richter, E., Niemann, S., Rüsch-Gerdes, S., and Hoffner, S. (1999). Identification of Mycobacterium kansasii by using a DNA probe (AccuProbe) and molecular techniques. J. Clin. Microbiol. 37, 964–970.
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Ricketts, W. M., O'Shaughnessy, T. C., and van Ingen, J. (2014). Human-to-human transmission of Mycobacterium kansasii or victims of a shared source? Eur. Respir. J. 44, 1085–1087. doi: 10.1183/09031936.00066614
Ross, B. C., Jackson, K., Yang, M., Sievers, A., and Dwyer, B. (1992). Identification of a genetically distinct subspecies of Mycobacterium kansasii. J. Clin. Microbiol. 30, 2930–2933.
Saini, V., Raghuvanshi, S., Talwar, G. P., Ahmed, N., Khurana, J. P., Hasnain, S. E., et al. (2009). Polyphasic taxonomic analysis establishes Mycobacterium indicus pranii as a distinct species. PLoS ONE 4:e6263. doi: 10.1371/journal.pone.0006263
Saitou, N., and Nei, M. (1987). The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4, 406–425.
Santin, M., and Alcaide, F. (2003). Mycobacterium kansasii disease among patients infected with human immunodeficiency virus type 1: improved prognosis in the era of highly active antiretroviral therapy. Int. J. Tuberc. Lung Dis. 7, 673–677.
Santos, A. R., De Miranda, A. B., Lima, L. M., Suffys, P. N., and Degrave, W. M. (1992). Method for high yield preparation in large and small scale of nucleic acids from mycobacteria. J. Microbiol. Methods 28, 1236–1243.
Shaaban, H., Layne, T., Sensakovic, J. W., and Boghossian, J. (2014). Mycobacterium kansasii septicaemia in an AIDS patient complicated by acute respiratory distress syndrome and acute liver failure. Int. J. STD AIDS 25, 152–154. doi: 10.1177/0956462413496080
Shahraki, A. H., Trovato, A., Mirsaeidi, M., Borroni, E., Heidarieh, P., Hashemzadeh, M., et al. (2017). Mycobacterium persicum sp. nov., a novel species closely related to Mycobacterium kansasii and Mycobacterium gastri. Int. J. Syst. Evol. Microbiol. 67, 1766–1770. doi: 10.1099/ijsem.0.001862
Shitrit, D., Baum, G. L., Priess, R., Lavy, A., Shitrit, A. B., Raz, M., et al. (2006). Pulmonary Mycobacterium kansasii infection in Israel, 1999-2004: clinical features, drug susceptibility, and outcome. Chest 129, 771–776. doi: 10.1378/chest.129.3.771
Sood, G., and Parrish, N. (2017). Outbreaks of nontuberculous mycobacteria. Curr. Opin. Infect. Dis. 30, 404–409. doi: 10.1097/QCO.0000000000000386
Tagini, F., Aeby, S., Bertelli, C., Droz, S., Casanova, C., Prod'hom, G., et al. (2019). Phylogenomics reveal that Mycobacterium kansasii subtypes are species-level lineages. Description of Mycobacterium pseudokansasii sp. nov., Mycobacterium innocens sp. nov. and Mycobacterium attenuatum sp. nov. Int. J. Syst. Evol. Microbiol. 69, 1696–1704. doi: 10.1099/ijsem.0.003378
Taillard, C., Greub, G., Weber, R., Pfyffer, G. E., Bodmer, T., Zimmerli, S., et al. (2003). Clinical implications of Mycobacterium kansasii species heterogeneity: Swiss National Survey. J. Clin. Microbiol. 41, 1240–1244. doi: 10.1128/JCM.41.3.1240-1244.2003
Tanizawa, Y., Fujisawa, T., and Nakamura, Y. (2017). DFAST: a flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics 34, 1037–1039. doi: 10.1093/bioinformatics/btx713
Telenti, A., Marchesi, F., Balz, M., Bally, F., Böttger, E. C., and Bodmer, T. (1993). Rapid identification of mycobacteria to the species level by polymerase chain reaction and restriction enzyme analysis. J. Clin. Microbiol. 31, 175–178.
Thomson, R., Tolson, C., Carter, R., Coulter, C., Huygens, F., and Hargreaves, M. (2013). Isolation of nontuberculous mycobacteria (NTM) from household water and shower aerosols in patients with pulmonary disease caused by NTM. J. Clin. Microbiol. 51, 3006–3011. doi: 10.1128/JCM.00899-13
Thomson, R., Tolson, C., Huygens, F., and Hargreaves, M. (2014). Strain variation amongst clinical and potable water isolates of M. kansasii using automated repetitive unit PCR. Int. J. Med. Microbiol. 304, 484–489. doi: 10.1016/j.ijmm.2014.02.004
Tortoli, E. (2014). Microbiological features and clinical relevance of new species of the genus Mycobacterium. Clin. Microbiol. Rev. 27, 727–752. doi: 10.1128/CMR.00035-14
Tortoli, E., Fedrizzi, T., Meehan, C. J., Trovato, A., Grottola, A., Giacobazzi, E., et al. (2017). The new phylogeny of the genus Mycobacterium: the old and the news. Infect. Genet. Evol. 56, 19–25. doi: 10.1016/j.meegid.2017.10.013
Tortoli, E., Simonetti, M. T., Lacchini, C., Penati, V., and Urbano, P. (1994). Tentative evidence of AIDS-associated biotype of Mycobacterium kansasii. J. Clin. Microbiol. 32, 1779–1782.
Vaidya, G., Lohman, D. J., and Meier, R. (2011). SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27, 171–180. doi: 10.1111/j.1096-0031.2010.00329.x
van der Wel, N., Hava, D., Houben, D., Fluitsma, D., van Zon, M., Pierson, J., et al. (2007). M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 129, 1287–1298. doi: 10.1016/j.cell.2007.05.059
van Ingen, J., de Zwaan, R., Dekhuijzen, R., Boeree, M., and van Soolingen, D. (2009). Region of difference 1 in nontuberculous Mycobacterium species adds a phylogenetic and taxonomical character. J. Bacteriol. 191, 5865–5867. doi: 10.1128/JB.00683-09
Varghese, N. J., Mukherjee, S., Ivanova, N., Konstantinidis, K. T., Mavrommatis, K., Kyrpides, N. C., et al. (2015). Microbial species delineation using whole genome sequences. Nucleic Acids Res. 43, 6761–7671. doi: 10.1093/nar/gkv657
Velayati, A. A., Boloorsaze, M. R., Farnia, P., Mohammadi, F., Karam, M. B., Masjedi, M. R., et al. (2005). Mycobacterium gastri causing disseminated infection in children of same family. Pediatr. Pulmonol. 39, 284–287. doi: 10.1002/ppul.20186
Volkman, H. E., Pozos, T. C., Zheng, J., Davis, J. M., Rawls, J. F., and Ramakrishnan, L. (2010). Tuberculous granuloma induction via interaction of a bacterial secreted protein with host epithelium. Science 327, 466–469. doi: 10.1126/science.1179663
Witzig, R. S., Fazal, B. A., Mera, R. M., Mushatt, D. M., Dejace, P. M., Greer, D. L., et al. (1995). Clinical manifestations and implications of coinfection with Mycobacterium kansasii and human immunodeficiency virus type 1. Clin. Infect. Dis. 21, 77–85. doi: 10.1093/clinids/21.1.77
Wu, T. S., Leu, H. S., Chiu, C. H., Lee, M. H., Chiang, P. C., Wu, T. L., et al. (2009). Clinical manifestations, antibiotic susceptibility and molecular analysis of Mycobacterium kansasii isolates from a university hospital in Taiwan. J. Antimicrob. Chemother. 64, 511–514. doi: 10.1093/jac/dkp238
Yang, M., Ross, B. C., and Dwyer, B. (1993). Identification of an insertion sequence-like element in a subspecies of Mycobacterium kansasii. J. Clin. Microbiol. 31, 2074–2079.
Yoon, S. H., Ha, S. M., Lim, J. M., Kwon, S. J., and Chun, J. (2017). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie van Leeuwenhoek. 110, 1281–1286. doi: 10.1007/s10482-017-0844-4
Zhang, M., Chen, J. M., Sala, C., Rybniker, J., Dhar, N., and Cole, S. T. (2014). EspI regulates the ESX-1 secretion system in response to ATP levels in Mycobacterium tuberculosis. Mol. Microbiol. 93, 1057–1065. doi: 10.1111/mmi.12718
Keywords: Mycobacterium kansasii complex, Mycobacterium ostraviense sp. nov., non-tuberculous mycobacteria (NTM), whole genome sequencing, taxonomy
Citation: Jagielski T, Borówka P, Bakuła Z, Lach J, Marciniak B, Brzostek A, Dziadek J, Dziurzyński M, Pennings L, van Ingen J, Žolnir-Dovč M and Strapagiel D (2020) Genomic Insights Into the Mycobacterium kansasii Complex: An Update. Front. Microbiol. 10:2918. doi: 10.3389/fmicb.2019.02918
Received: 24 August 2019; Accepted: 04 December 2019;
Published: 15 January 2020.
Edited by:
Miguel Viveiros, New University of Lisbon, PortugalReviewed by:
Igor Mokrousov, Saint Petersburg Pasteur Institute, RussiaVikram Saini, All India Institute of Medical Sciences, India
Copyright © 2020 Jagielski, Borówka, Bakuła, Lach, Marciniak, Brzostek, Dziadek, Dziurzyński, Pennings, van Ingen, Žolnir-Dovč and Strapagiel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tomasz Jagielski, t.jagielski@biol.uw.edu.pl; Dominik Strapagiel, dominik.strapagiel@biol.uni.lodz.pl
†These authors have contributed equally to this work